Circular polymerase extension cloning (CPEC)
Tired of not being able to clone a CRISPR library w/ a Gini coefficient < 0.2? Well, outside of artificially shrinking your Gini estimate by log transforming the reads (@MaGeck), you can always try circular polymerase extension cloning! It’s way easier than restriction enzyme approaches and works like a charm for certain applications where Gibson assembly fails.
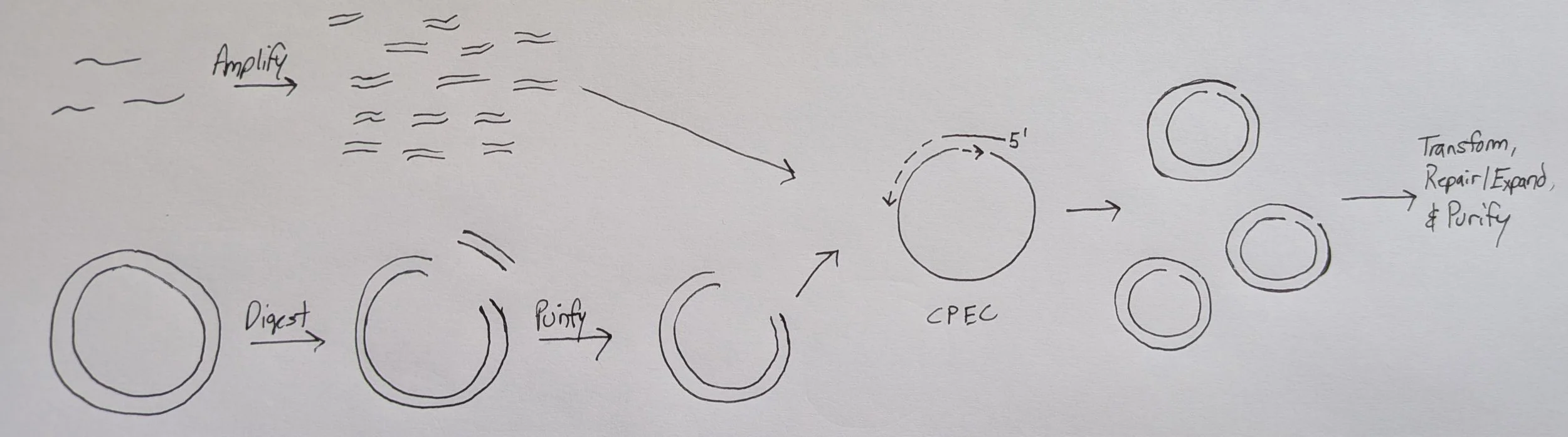
CPEC scarlessly inserts DNA fragments of variable sequence into a linearized backbone by annealing matching ends in a one-pot reaction similar to Gibson assembly except temperature is not held constant but fluctuated like in PCR. Furthermore, no exonuclease or ligase is included, only a high-fidelity polymerase to extend the annealed fragments. The rxn is cycled to linearly increase the # of successful assemblies with the final products (in our case) being circular dsDNA with two staggered nicks (one on each strand) aka floppy plasmids. These nicks are easily repaired upon transformation into bacteria. Essentially, it’s PCR using the inserts as individual “bridge” primers meaning you can run directly with whatever 2x PCR master mix (Phusion Plus, NEB Ultra, etc.) you have stocked in lab if you ever feel like testing out.
Also known as a “lazy gibson”. Exclude the specialty enzymes (T5 exo, Taq ligase) and let your bacteria do the final repair of the nicked product!
Shout out to this message board for introducing me to the method while I was troubleshooting my approach to cloning a CRISPRi library by Gibson:
Poor gibson efficiency in lenti CRISPR V2 - https://groups.google.com/g/crispr/c/ZWJF7iw7e90?pli=1
“But Gibson always works!” It’s widely great, but for some applications (like inserting spacers next to a scaffold region w/ lots of secondary structure) it fails. Learn why below!
Let’s start with Gibson…
Theoretically straightforward, great for cloning one-off constructs quickly & without hassle and stitching many fragments together at once, I figured why not try out for cloning a small (n=330 constructs) CRISPRi library?
Here’s the original paper from 2009 (10k+ citations): https://www.nature.com/articles/nmeth.1318
If you checked out my “cloning a CRISPR library” protocol you’ll see I did it the “old-fashioned” way via restriction digest followed by ligation. It worked but Gini of the final library was > 0.2 and working w/ such small DNA fragments (<30bp) was a hassle. Therefore, I was curious how Gibson would compare and ordered a similar library of spacers from TWIST w/ end regions designed for Gibson cloning into a lntCRISPRv2 plasmid derivative built for CRISPRi (swapped in ZIM3-dCas9)
I like the Gibson master mix from NEB even though it’s pricey b/c it just works: NEB Hifi 2x Assembly Mix ($735 for 50 rxns)
At its core, a Gibson rxn contain a 5’ exonuclease (for chew back), a polymerase (for fill in), and a DNA ligase (for sealing) in a compatible buffer w/ all necessary cofactors (e.g. deoxynucleoside triphosphates) and PEG 8000 to encourage interactions
Usually Taq ligase is used b/c of it’s thermostability (uses NAD+ as cofactor & only seals nicked ends, not blunt, unlike T4 ligase)
T5 exonuclease is used specifically b/c it’s highly unstable at 50C so it only works in the first few minutes of the rxn to partially chew back the DNA
Usually Phusion or Q5 is used as the polymerase b/c they’re high fidelity (proofreading) and non strand displacing
If ~$15/rxn is too much for you (fair), you can halve or quarter the total rxn volume to 5-10uL. Alternatively, here’s a homebrew Gibson 2x MM recipe:
https://wikis.mit.edu/confluence/display/LULAB/Gibson+Assembly+Master+mix+preparation
Typically, you want 15-30bp of overlap between end regions of the backbone and insert(s) you’re trying to covalently combine
The rxn is run at 50C to balance the activity of all enzymes while ensuring complementary DNA overhangs can anneal (this will be important later)
Note: the schematic on the right shows the DNA starting w/ blunt ends but it can also have staggered/sticky ends
5’ or 3’ overhangs, hydroxylated or phosphorylated, honeybadger (T5 exo) don’t care, which is another reason why it’s used in Gibson assemblies
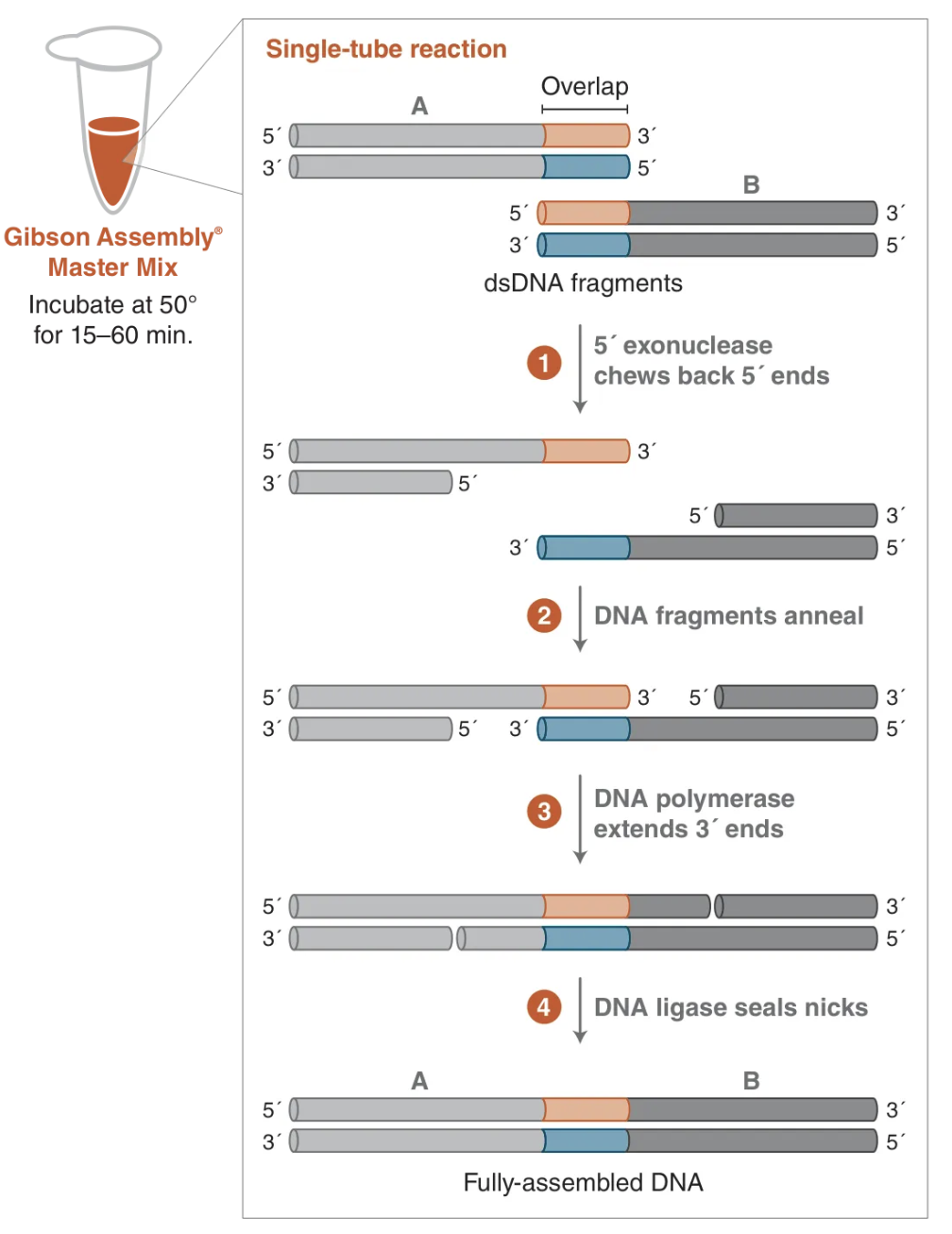
Image from NEB
Oligo design for Gibson assembly of CRISPRi library:
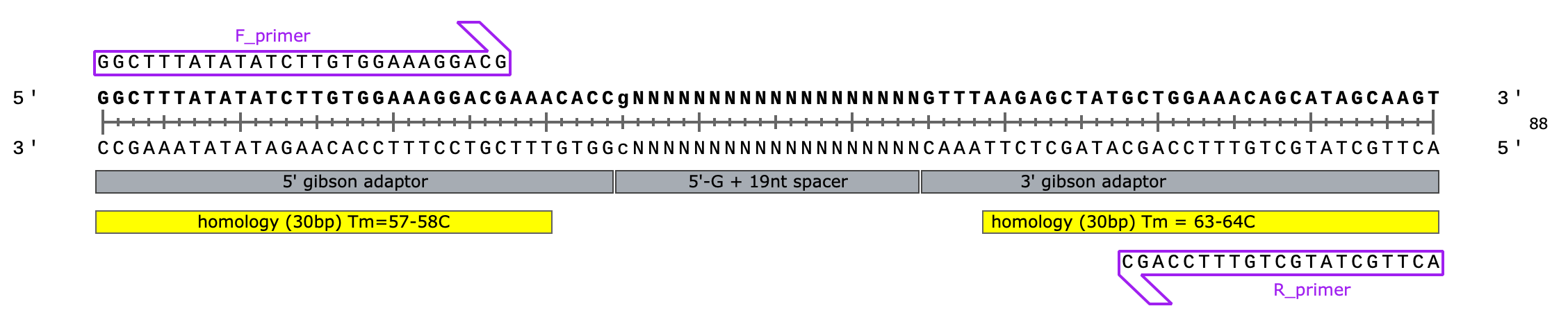
spacer: 5'- gNNNNNNNNNNNNNNNNNNN - 3' (length = 20 nt)
Leftside adaptor) 5’ - GGCTTTATATATCTTGTGGAAAGGACGAAACACC - 3’ (34 nt total, 30 nt overlap)
This is the end of the U6 promoter, immediately upstream of the sgRNA in the final plasmid design
Rightside adaptor) 5’ - GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGT - 3’ (34 nt total, 30 nt overlap)
This is the start of the sgRNA scaffold, immediately downstream of the targeting spacer seq. we’re inserting into the plasmid
34 +20 +34 = 88 nt oligo library (cheap/easy to synthesize)
88-30-30=28bp non-overlapping (expected Tm = 60-70C)… i.e. should hold together during 50C assembly
This is important & pushing the limits, usually Gibson assembly of inserts <100 bp in length is not recommended but it’s doable if you keep rxn time short (<=15 min) and increase the molar ratio of fragment:backbone in the rxn (I recommend 5:1 or 10:1)
The reason short (<100bp) inserts are discouraged is that T5 exo works fast at the start of the rxn and there’s a possibility your whole insert fragment is chewed through before T5 is inactivated. Or chewed enough that whatever dsDNA remains melts apart
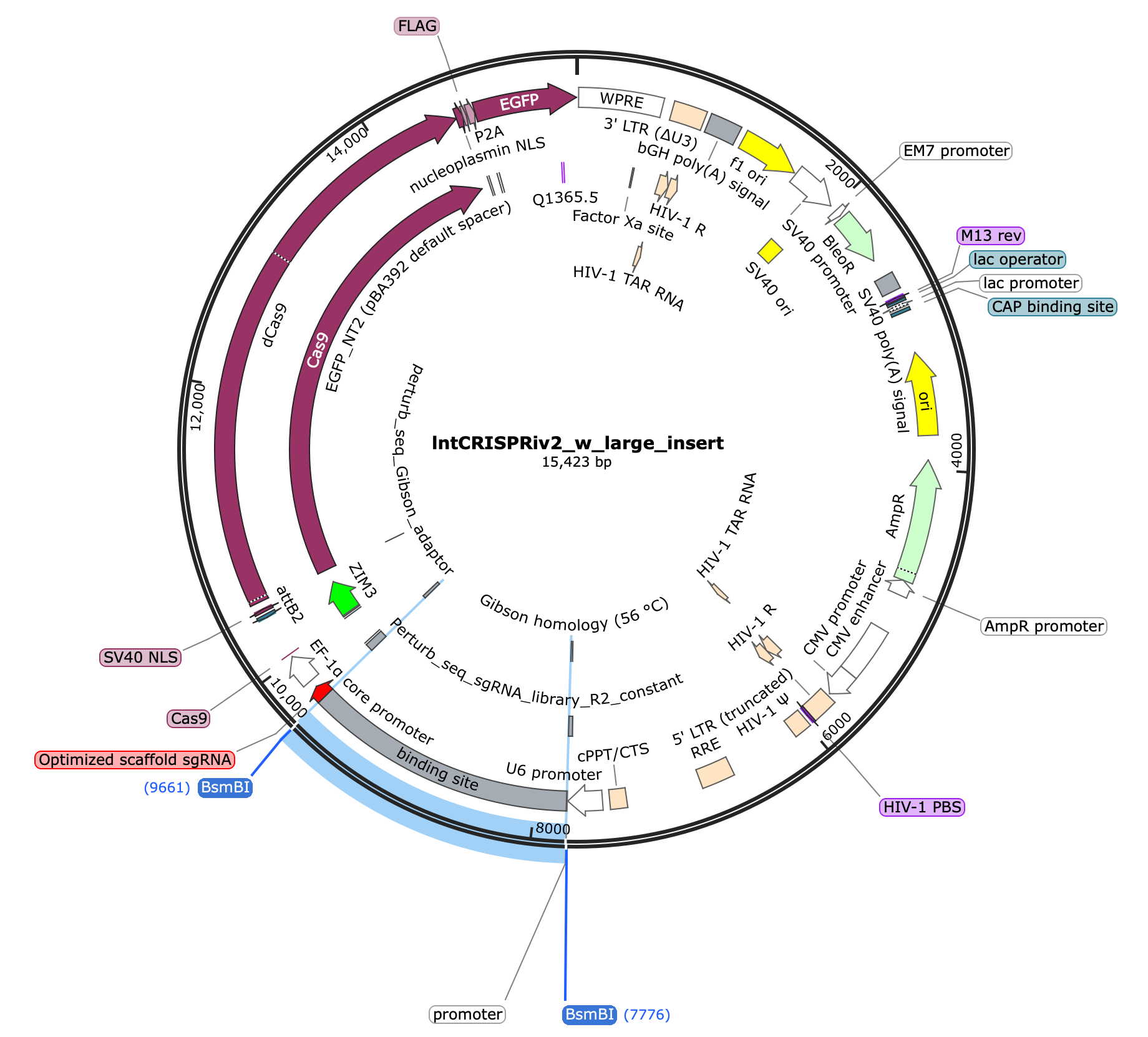
lntCRISPRiv2_w_large_insert plasmid (pre-digestion) 15,423 bp
lntCRISPRiv2 linearized backbone (post-digestion) 13,542 bp
Oligo library: 88bp
Final product length is 13,542 + 20 (spacer length i.e. the only new sequence added) = 13,562 bp
PCR primers for library amplification once oligos arrive:
“F_primer”) 5’ - GGCTTTATATATCTTGTGGAAAGGACG - 3’ (Tm = 57-58C, length = 27nt)
“R_primer”) 5’ - ACTTGCTATGCTGTTTCCAGC - 3’ (Tm = 58-59C, length = 21nt)
Digest with BsmBI to linearize
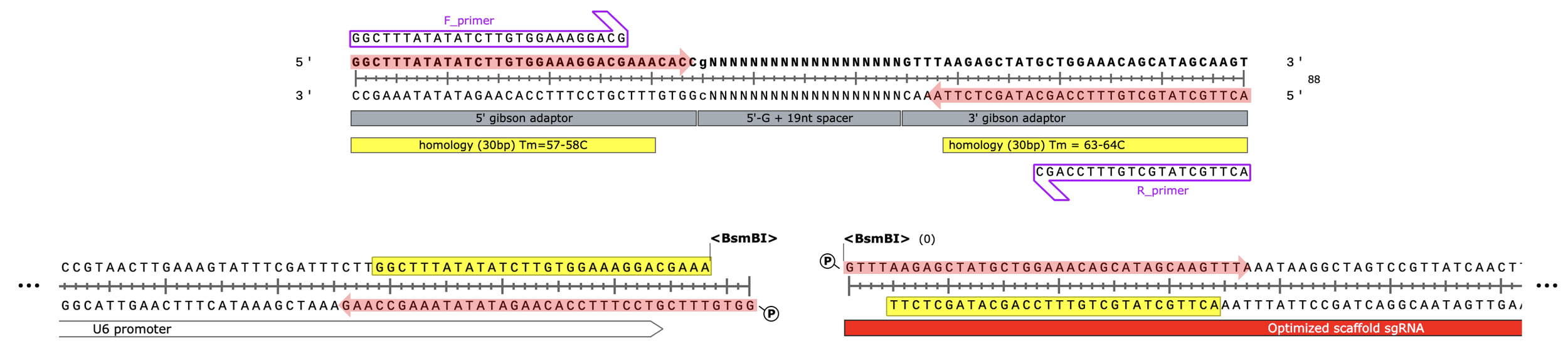
Amplified (v3) spacer library:
BsmBI-v2 digested backbone:
Red shaded arrows indicate regions chewed back by T5 exonuclease during assembly
Of course, before we assemble, we must amplify our synthesized oligo pool by PCR:
v3 oligo pool (designed for CRISPRi via gibson assembly) from TWIST arrived with 56 ng total
Spin to 15,000g to settle lyophilized DNA at bottom of tube
Resuspend with 18 uL dH2O. Vortex, spin down, and incubate to get DNA into soln.
56/18 = 3.11 ng/uL
Resuspend PCR0 primers to 100 uM w/ dH2O
v3 F_primer
v3_R_primer
Prepare PCR master mix (enough for n=10 rxns)
25 uL * 10 = 250 uL Phusion Plus
23 uL * 10 = 230 uL dH2O
0.25 uL *10 = 2.5 uL of F_primer (100 uM)
0.25 uL *10 = 2.5 uL of R_primer (100 uM)
Run a 2 rxn PCR test
Rxn 1) 48 uL MM + 3 uL library
Rxn 2) 48 uL MM + 3 uL dH2O
5 cycles total
Thermal conditions:
98C for 30s
[98C, 10s —> 60C, 10s —> 72C, 20s]*5 cycles
72C, 2 min
4C, hold
Dilute completed PCR rxns 1:20 and 1:10 in dH2O and run on 4% agarose (EZ)gel for 10-15 min
Included 100bp and 10bp ladders in leftmost lanes
Once band confirmed, repeat PCRs using remainder of starting material (n=6 rxns total)
Pool all 6 positive rxns for a total of ~300 uL & purify by SpriSelect
Add 300 uL SpriSelect beads then add 300 uL isopropanol
Wash 2x85% EtOH.
Elute in 33 uL dH2O, transferring 32 uL to new tube. Measure by Nanodrop
33.7 ng/uL (1.8/2.1) ----33 uL----> 1,089 ng total
Theoretically, 56ng*(2^5) = 1,792 ng total
Once more, to be thorough, dilute small aliquot of amplified & concentrated v3 library and run on 4% agarose gel along w/ 10bp ladder (leftmost lanes)
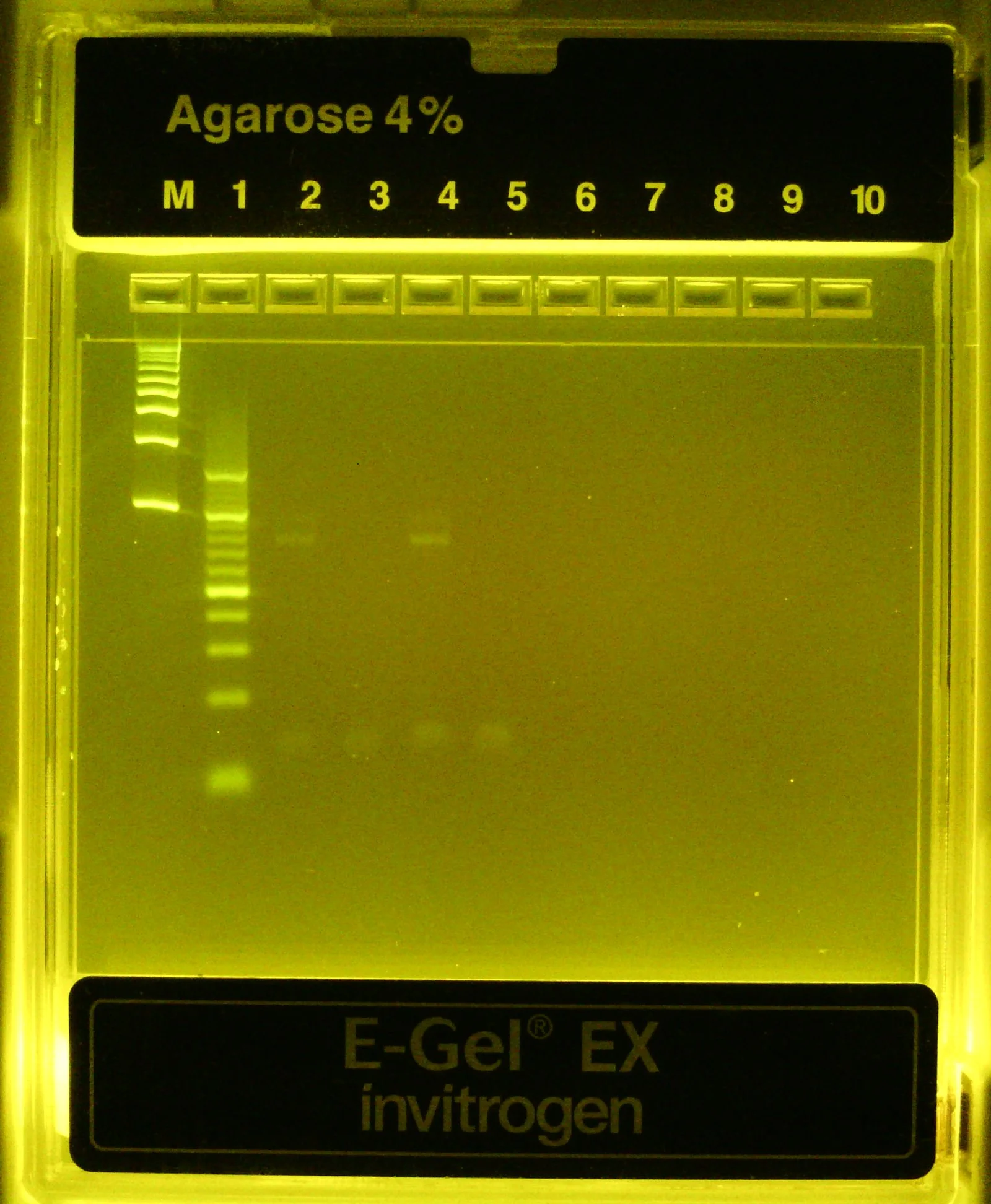
Expected band size = 88bp in lanes (2) and (4) only
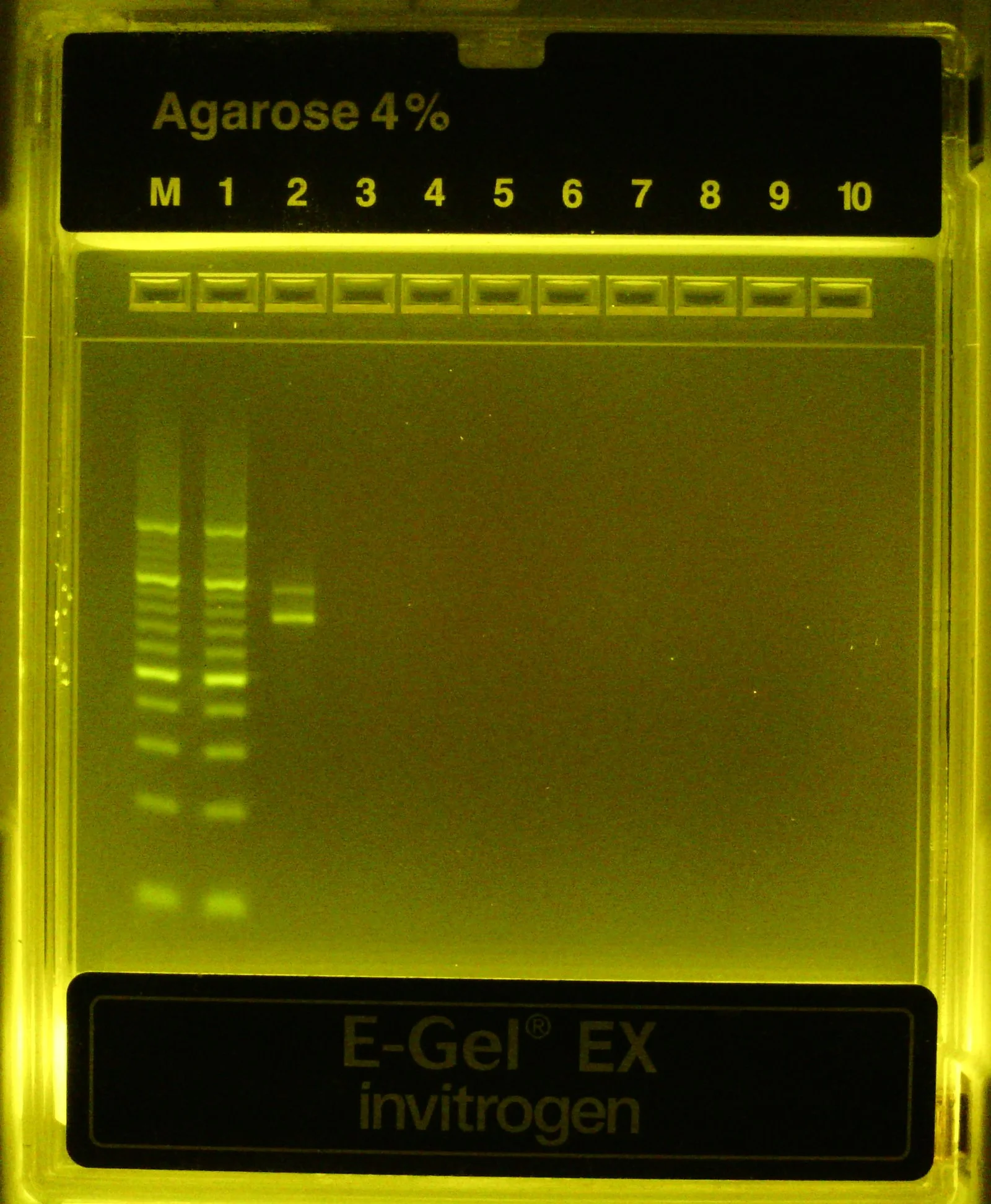
Huh, that’s weird, at higher resolution it appears we have a band at ~80bp and another at ~100 bp instead of a single band at 88bp…
Okay that’s odd, it appears we’re getting two bands from our PCR amplification of the oligo pool, neither appearing to be the exact size we expected (88bp). Let’s submit an aliquot of purified PCR product to Plasmidsaurus to verify by Premium PCR
Again, not sponsored, but if you ever need to troubleshoot a sample of linear dsDNA I highly recommend the Premium PCR option from Plasmidsaurus. For $30 / sample they’ll return up to 5,000 (usually 1,000-3,000) Nanopore-based long sequencing reads of whatever dsDNA is in the tube - both Watson and Crick strands.
In their words, the “Premium PCR” sample sequencing service is “Amplification-free long-read sequencing with no fragmentation - ideal for sequencing PCR products when working with mixed populations of molecules, or when sufficient read depth or full length reads are required”.
Of course the reads are still noisy (base error rate ~= 1 in 100) but it’s a very helpful method for getting an initial sense of what’s in the tube. For better or worse, it only reads dsDNA (e.g. primers won’t show up) since they add the adaptors for ONT sequencing to any exposed, blunt ended dsDNA
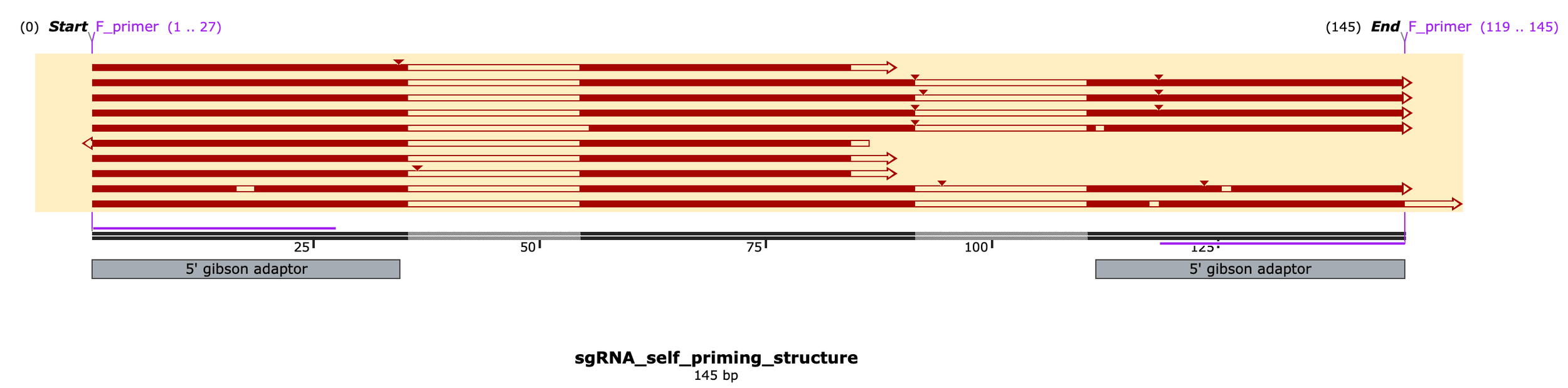
Okay so the results are very interesting… Looks like the primary peak of reads corresponds to the intended 88bp product while the secondary band (~145bp) is a construct that self-primed off the main sgRNA scaffold hairpin to include the reverse complementary sequence... wow
I knew there was risk in ordering oligos w/ known secondary structure (the partial sgRNA scaffold sequence) but just hoped it wouldn’t bite me and here it is biting me
So the correct product is 88bp dsDNA which i think actually corresponds to the TOP band on the gels above, just running a little slowly, whereas the lower band I think corresponds to the unanticipated self-primed hairpin product (145nt ssDNA folded over to make a pseudo 77bp dsDNA fragment w/ a single blunt end, hence why it was picked up by ONT sequencing & read in only one direction). Here a subset of 10 reads aligned to the reference sequence:
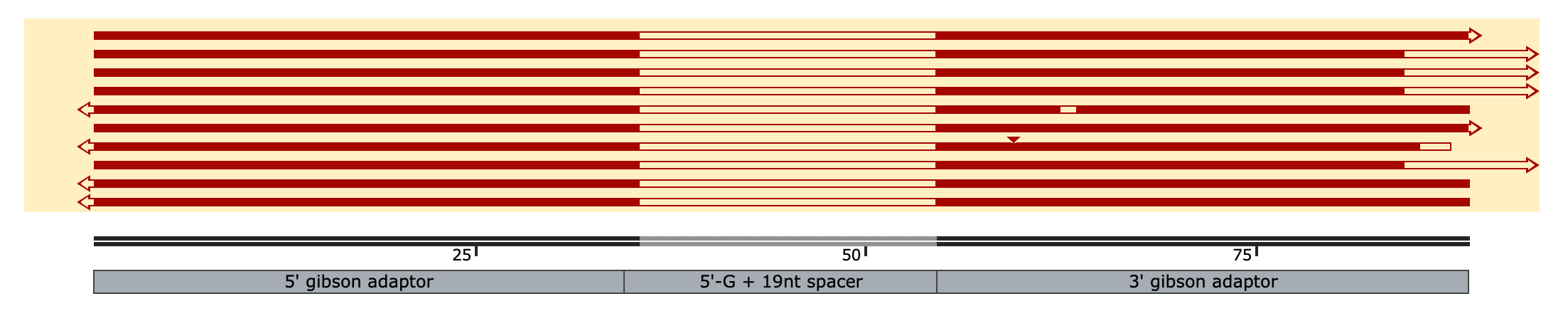
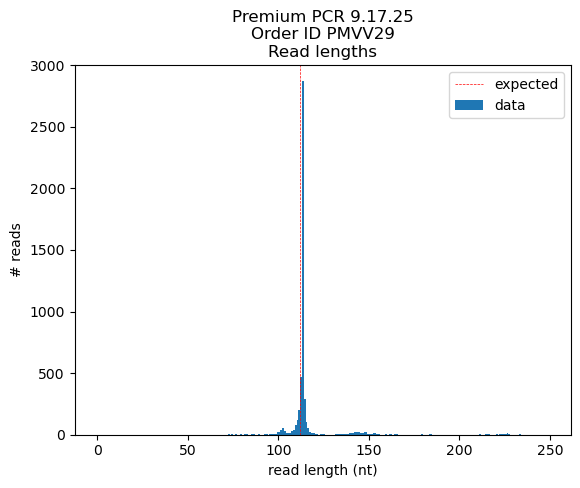
~40% of aligned reads (n=10 here, 5000 in total) extend much farther past the 3’ end of the reference seq, the rest match perfectly or near perfectly
(the middle un-matching regions are b/c of the different spacers in the pool whereas the short, 1 nt overhangs are an artifact from sequencing)
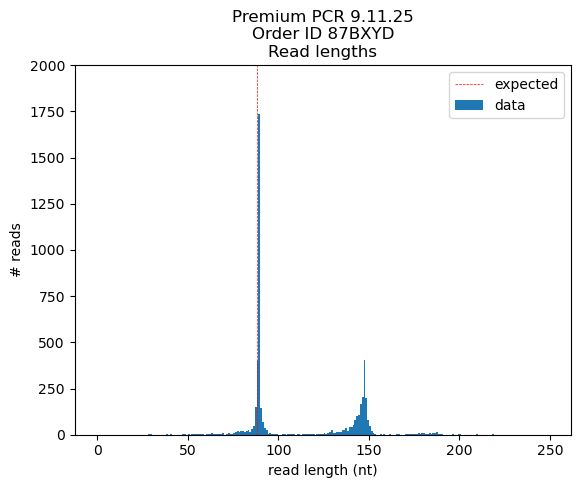
# < 100 nt: 2941 reads (est. peak size = 89 nt)
# > 100 nt: 2058 reads (est. peak size = 146 nt)
~90% of reads < 100 nt match or closely match the intended oligo structure:
# forward stranded) 1285 reads
# reverse stranded) 1342 reads
~70% of reads > 100nt match or closely match the self primed hairpin reference sequence:
# forward stranded) 1483 reads
# reverse stranded) 1 reads
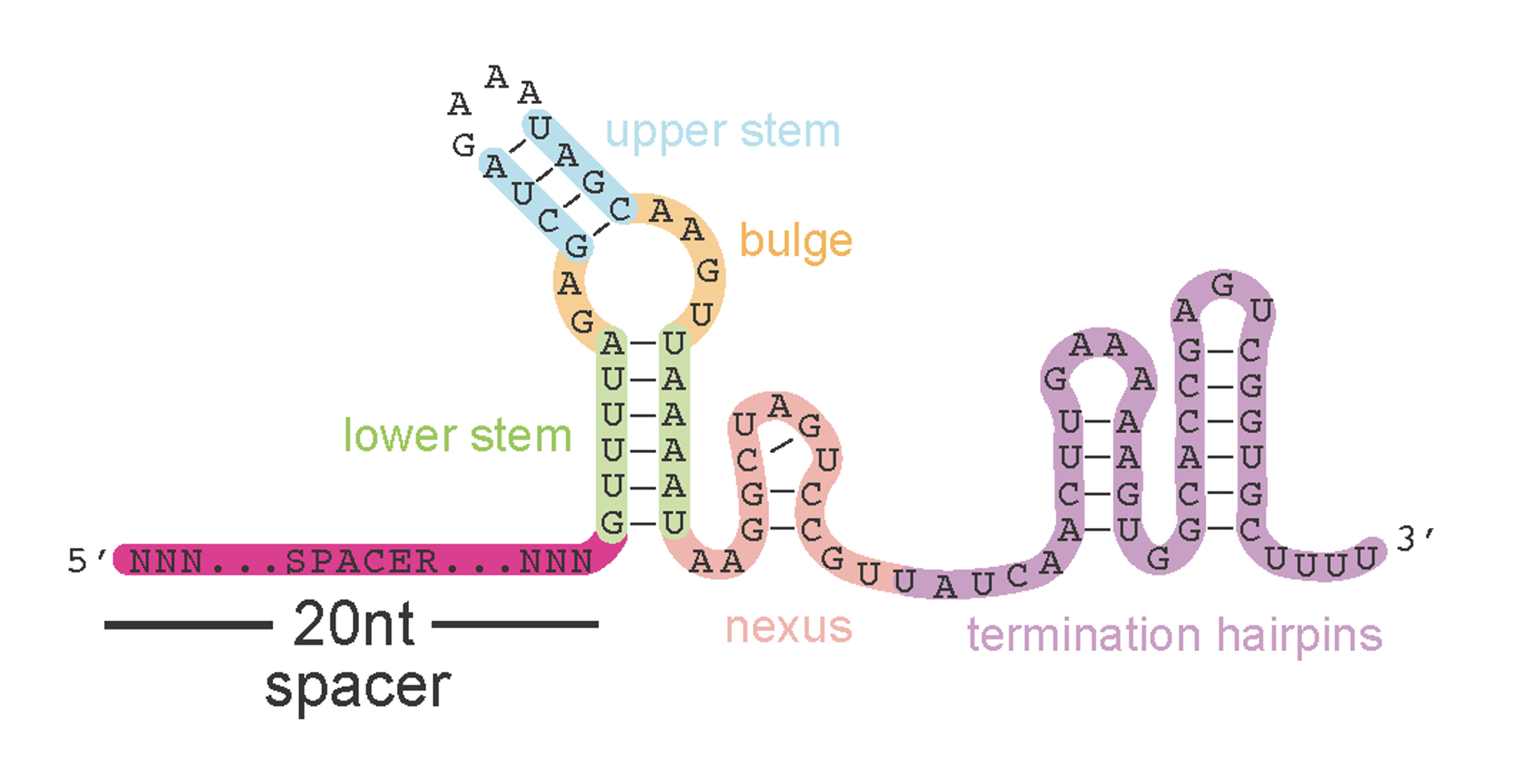
Engineered SpCas9 sgRNA secondary structure prediction #1
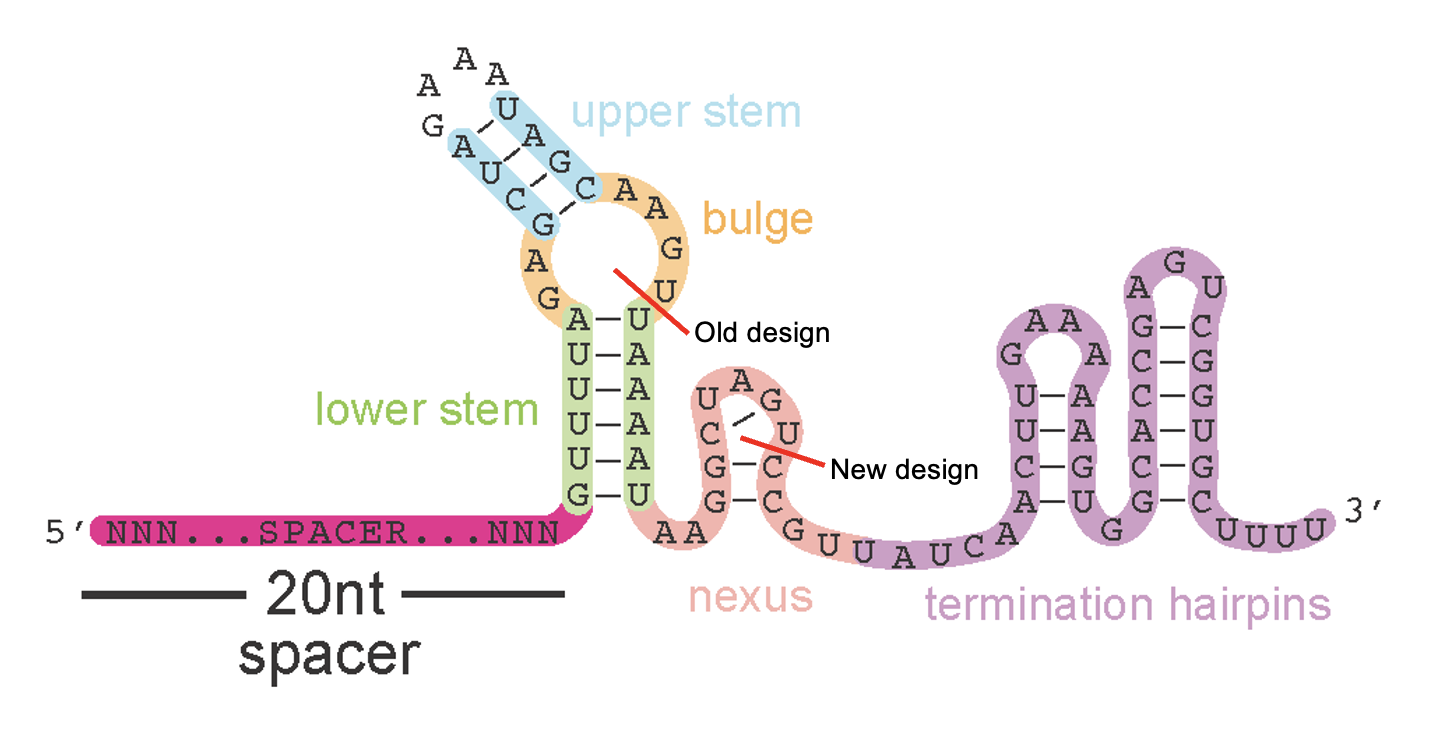
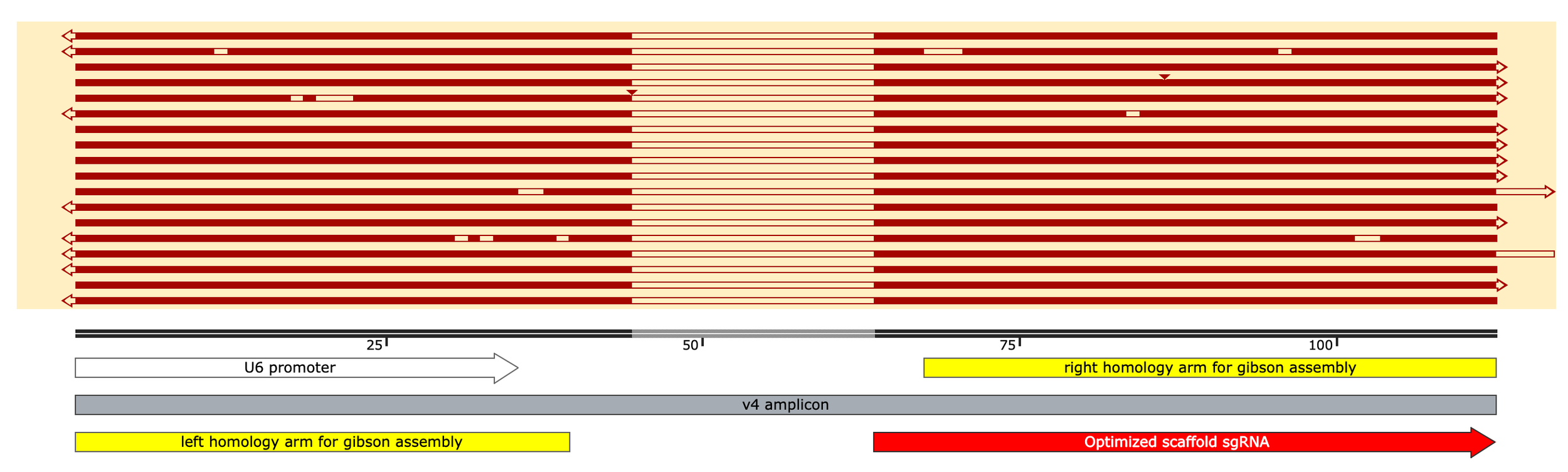
Okay so what’s going on in the 40% of reads that don’t match? A closer look reveals that these reads have two variable spacer regions and ends that both match the leftside (5’ gibson adaptor) sequence but in reverse complement of each other. How this happens starts to make sense if you look at the predicted secondary structure of the sgRNA scaffold sequence which our oligos contain part of, as their rightside adaptor, to anneal correctly into the backbone
A reminder on how the spCas9 sgRNA we’re using was optimized for in vivo expression & editing in mammalian cells circa 2012…
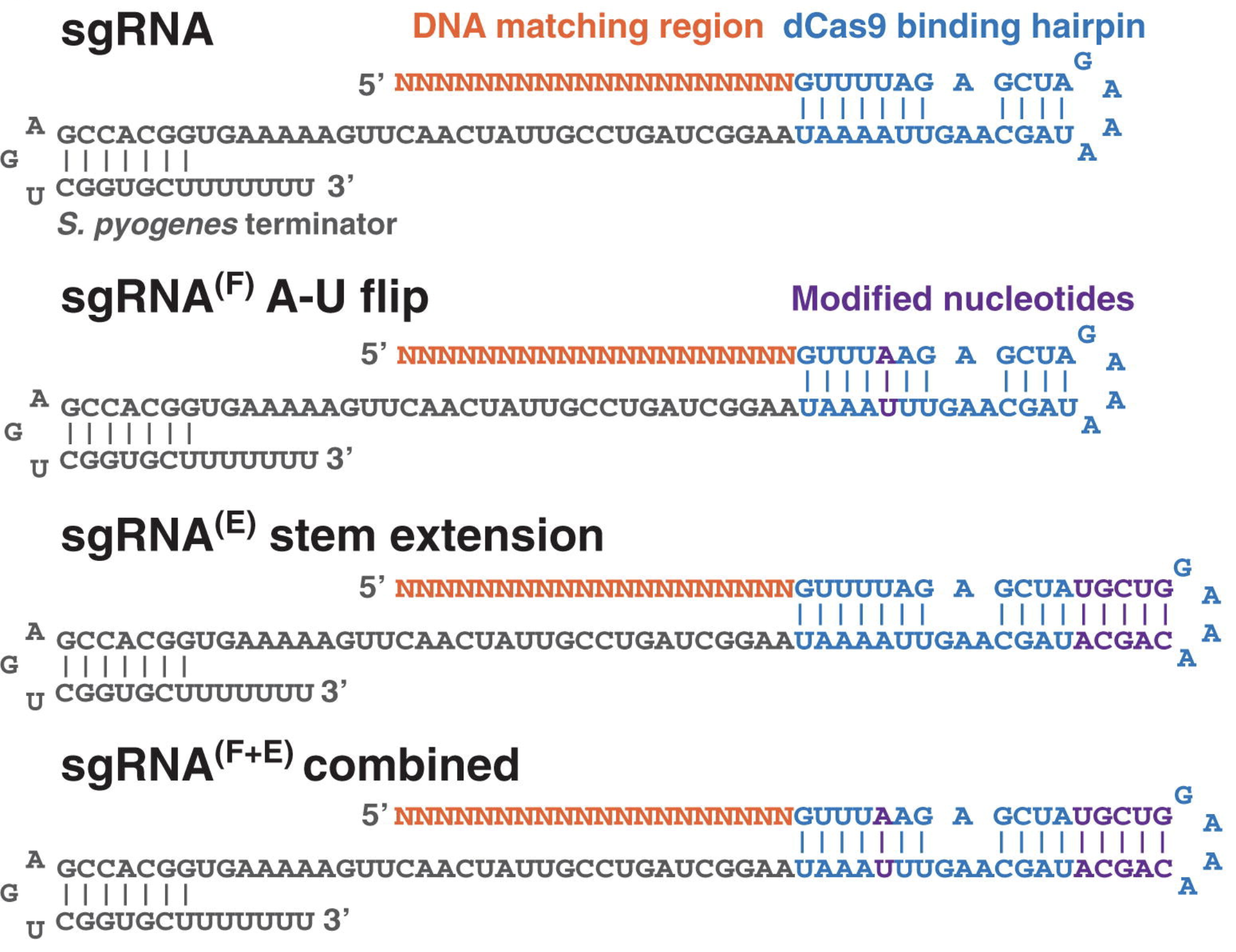
The oligos I ordered use this scaffold design and end around here
Chen, Baohui et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell, Volume 155, Issue 7, 1479 - 1491 (2013)
With our oligo design, during PCR amplification, the partial scaffold sequence can fold over to form the primary sgRNA hairpin and prime itself for “reverse extension” by proofreading polymerase after it chews back the 4nt overhang
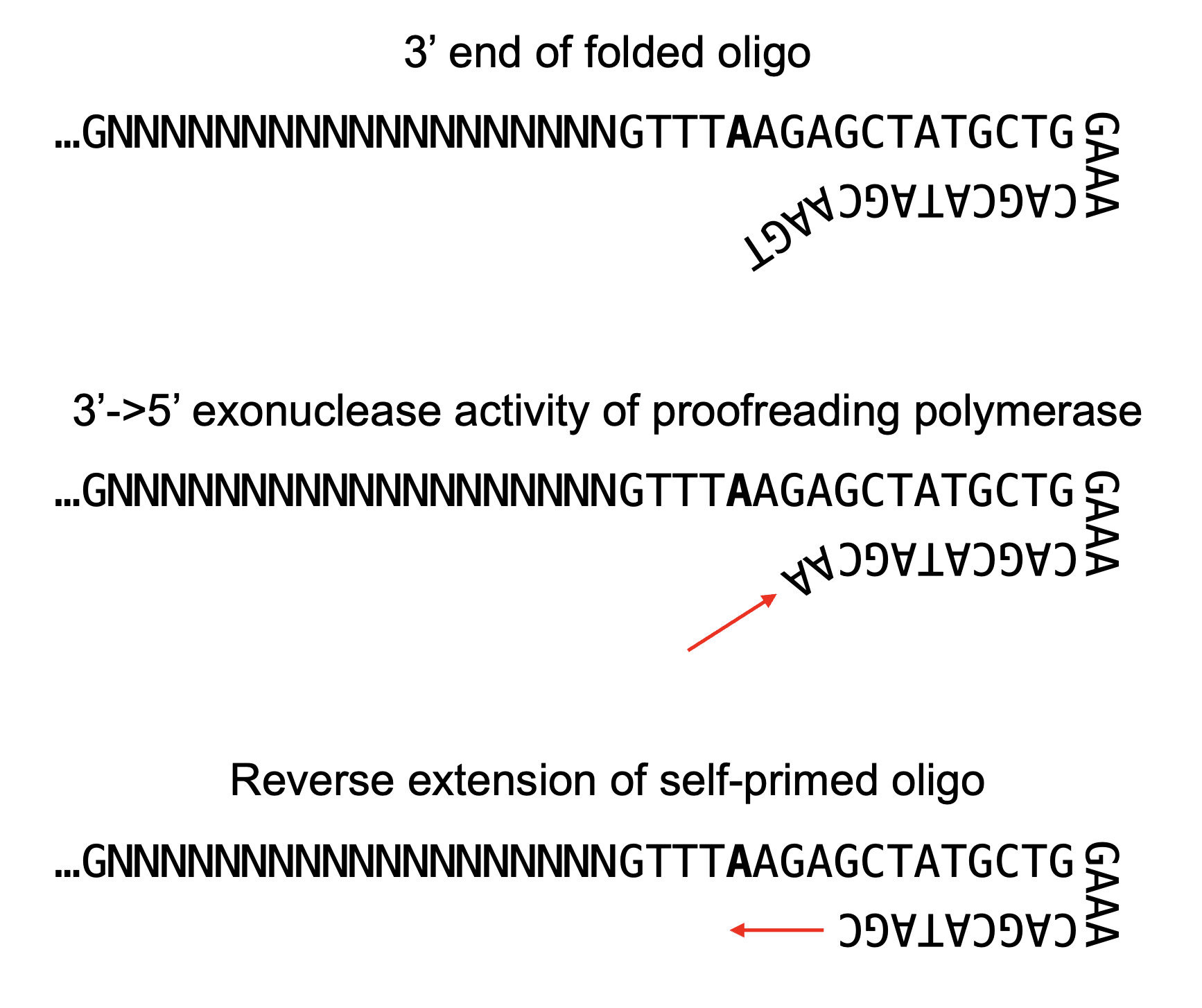
Can proofreading polymerases really chew back a 4nt long 3’ overhang? Yup, I talked to NEB about this and they suggested these enzymes can chew back up to 6-8nt overhangs, which the polymerase essentially views as a long replication error. We often think of polymerase proofreading as acting on one bad nucleotide immediately after misincorporation but that’s evidently not always the case.
As final proof of my proposed mechanism, self-primed constructs that were reverse extended should include a distinct “TCTTAAA“ sequence and align to the following reference map:
To the right, I subsample 10 reads and align to a reference sequence of the reverse extended self-primed construct (which would be 145 nt ssDNA aka ~77bp folded dsDNA once fully reverse extended, not 145 bp as indicated here by Snapgene)
Below, I zoom in on only reads that nearly perfectly align to this byproduct reference, note the “TCTTAAA” sequence and two, reverse complementary spacer regions confirming my theory

Okay, so clearly it’s not ideal to PCR a library of oligos with lots of secondary structure… Now this could also be because I used Phusion Plus MM for the PCR which encourages strand annealing by including a bunch of crowding agents in the buffer to achieve a “universal” annealing temp of 60C. To be safe, however, I re-designed and re-ordered the library of CRISPRi spacers for Gibson assembly so that the 3’ overhang of the folded hairpin structure is 9nt instead of 4nt hopefully rendering it unable to be chewed back by Phusion.
I also extended the 5’ end, now each homology arms shares 40-50 nt of sequence with each end of the backbone (a bit long for Gibson but should still work)
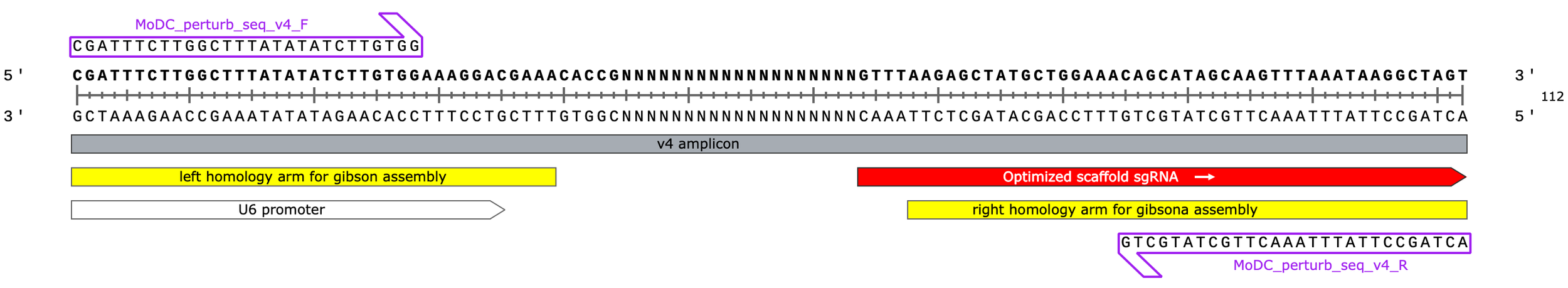
v4 oligo design
(we started with v3, the other versions were for cloning a CRISPRko library by restriction digest)
Note: I’ve shown you now a couple different folding configurations (secondary structure predictions) for the spCas9 sgRNA scaffold region. Both formations are possible but I think the one below on the right, which includes additional, smaller hairpins at the 3’ end, is most accurate. In this more-folded configuration, it’s very tricky to find a rightside homology arm for gibson assembly that doesn’t (1) leave a short exposed end that can be chewed back in PCR and (2) end directly within a hairpin - either of which would lead to reverse extension and a secondary, undesirable PCR product. I think I found at least one good spot, however, that meets the criteria. The other option? Extend the oligo design past the entire scaffold region however that’s $$$

Does the new design work (i.e. PCR amplify cleanly)?
Received MoDC perturb seq v4 (for CRISPRi via gibson cloning) oligo pool from TWIST… 39 ng total
Spin to 15,000g to settle lyophilized DNA at bottom of tube
Resuspend with 14 uL dH2O. Vortex, spin down, and incubate to get DNA into soln.
39/14=2.786 ng/uL
Re-suspend PCR0 primers to 100 uM w/ dH2O
MoDC_perturb_seq_v4_F
MoDC_perturb_seq_v4_R
Prepare PCR master mix (enough for n=6 rxns)
25 uL * 6 = 150 uL NEB Ultra II (notice the switch from Phusion Plus)
22 uL * 6 = 132 uL dH2O
0.25 uL *6 = 1.5 uL of F_primer (100 uM)
0.25 uL *6 = 1.5 uL of R_primer (100 uM)
Run a 2 rxn test PCR
Rxn 1) 48 uL MM + 3.3 uL library
Rxn 2) 48 uL MM + 3.3 uL dH2O
6 cycles total
Thermal conditions:
98C for 30s
[98C, 10s
64C, 20s
72C, 30s]*6 cycles
72C, 2 min
4C, hold
Lanes (1) and (3) are diluted PCR product from the same rxn that received template. Lanes (2) and (4) are from a no template control PCR rxn
Outer lanes are a 10bp DNA ladder
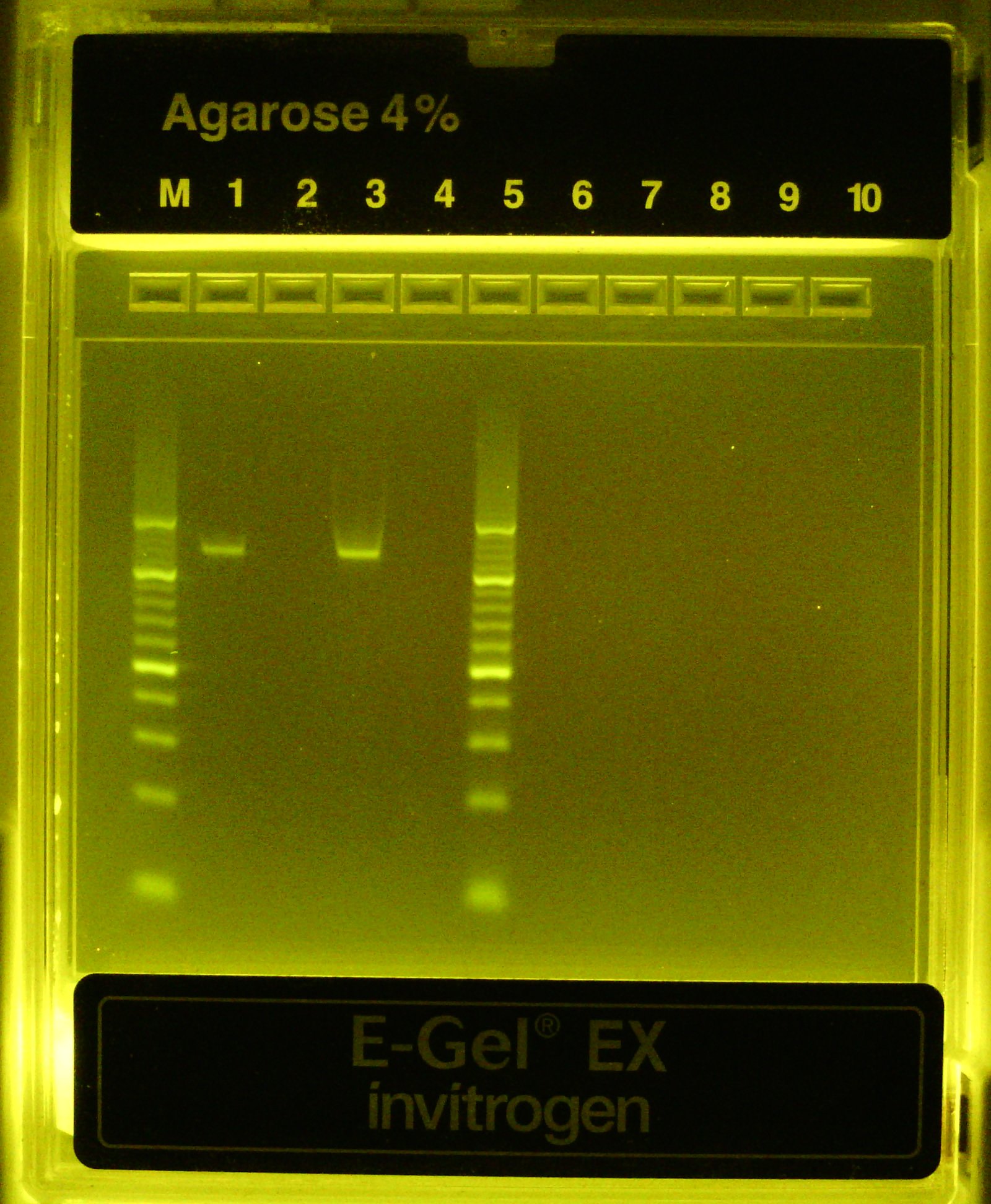
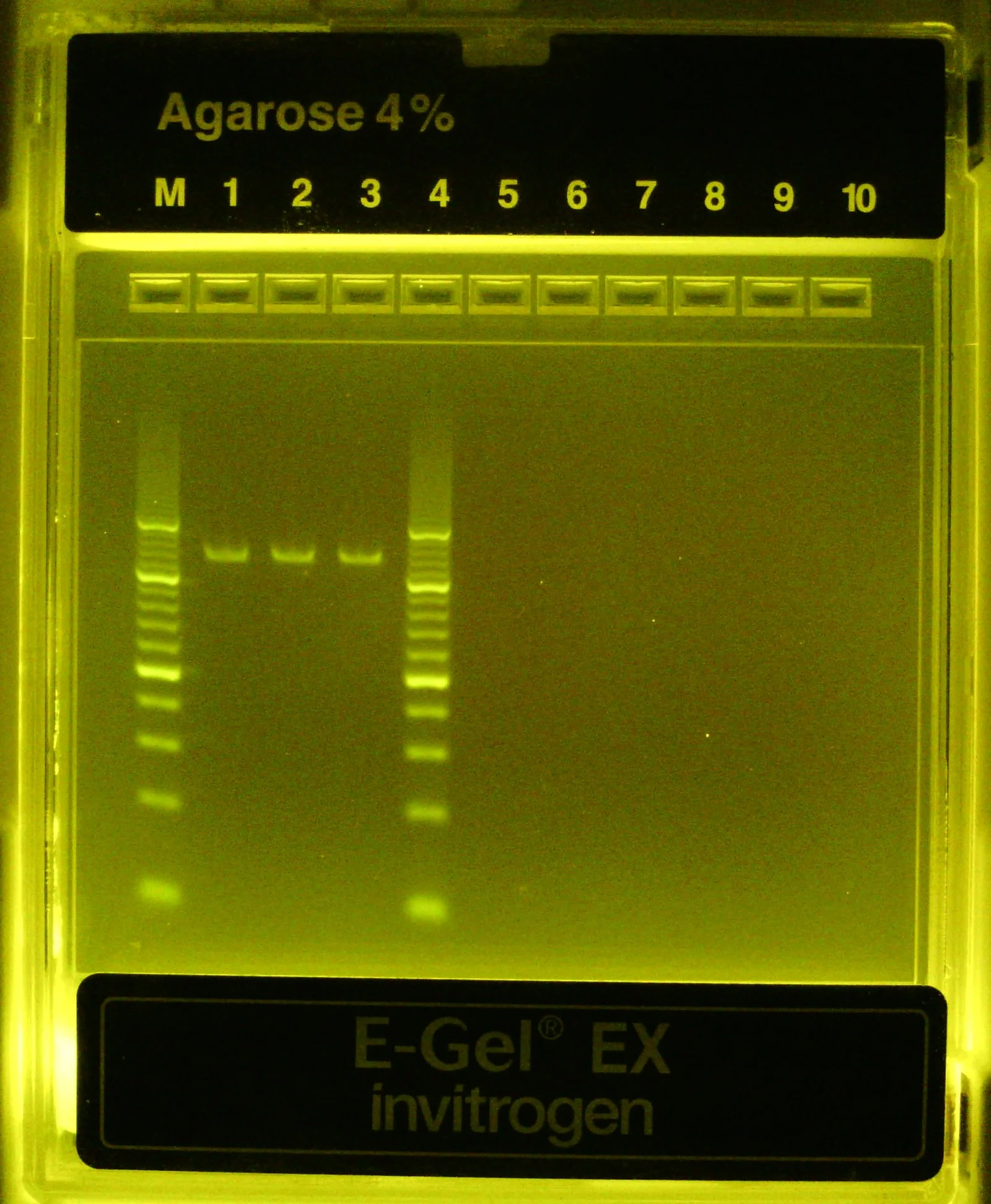
No negative controls this time, all 3 PCR rxns successful
Once finished (~15 min), dilute PCR product 1:20 and 1:10 into 20 uL aliquots and run on 4% agarose (EZ)gel for 15 min w/ 10 bp ladder
Test PCRs look successful! Single band at ~110 bp only when template is present (expected amplicon size = 112 bp)
Proceed to amplify all remaining material
Spike remaining ~10 uL of v4 oligo pool (2.8 ng/uL) into 150 uL of 1X PCR master mix (same as used for test rxn above)
Split 3x50 uL
Run w/ same thermocycler settings
Once completed, dilute PCR rxns 1:20 and run on 4% agarose (EZ)gel - confirmed successful
Pool all 4 positive rxns for a total of ~190 uL
Add 190 uL SpriSelect beads and then 100 uL isopropanol.
~22% final isopropanol concentration should be enough to precipitate our 112bp amplicon
Wash DNA-bound beads 2x with 85% EtOH.
Elute DNA in 55 uL dH2O, transferring 52 uL to new tube.
Measure by Nanodrop
Amplified v4 insert library) 33 ng/uL (1.8/2.0) ---*55 uL----> 33*55 = 1.8 ug (i.e. we amplified our total material ~45x)
The #s in parentheses are the 260nm/280nm and 260nm/230nm sample absorbance ratios, respectively, an indication of nucleic acid purity
What should we have expected in terms of yield with 100% PCR efficiency?
40*(2^6)=2,560 ng total (vs.1,800 ng observed i.e. our amplification+purification appears to have been 70% efficient overall… not bad)
Much cleaner this time around, yay! Most reads are the expected size and align well to the reference sequence (designed structure)
A random subset of sequencing reads aligned to the reference seq for oligo design v4
Final check
Dilute 1 uL of amplified v4 library into 30 uL dH2O in strip tube and submit to Plasmidsaurus for Premium PCR
Recommended submission concentration for ~100bp fragments = 1 ng/uL
33*(1/31)=1.0645 ng/uL submission concentration
Okay great, our pool of CRISPRi spacers w/ compatible ends is ready for assembly.
I’m going to pass over how to linearize the backbone, if you’re interested check out my “Cloning a CRISPR library” protocol. Briefly, we treat the lntCRISPRv2 derived plasmid w/ BsmBI-v2 for 1hr at 55C, purify/concentrate the digested DNA by Spri, run the product on a gel to confirm double digestion (both sites cut), then gel extract the primary band & purify once more by Spri.
With NEB Hifi 2x MM, we’re now all set for Gibson assembly.
Gibson assembly protocol:
Thaw NEB Hifi 2x MM on ice and NEBuilder positive control at room temp.
Prewarm thermocycler to 50C, warm 1 mL of LB to room temp.
In PCR strip tube, combine DNA fragments (e.g. 2.2 uL of insert + 3 uL linearized backbone, as indicated above) and dilute up to 10 uL w/ dH2O
For control rxn, transfer 10 uL of NEBuilder positive control to tube
Add 10 uL of 2x Hifi to each sample. Pipette to mix.
Now 20 uL total / tube
Seal tubes tightly and spin down briefly to settle liquid at bottom of tube.
Store on ice if still waiting for thermocyler to reach 50C
Incubate rxns 50°C for 10-20 minutes (when 2 or 3 fragments are being assembled) or 30-60 minutes (when 4–6 fragments are being assembled).
Immediately, transfer completed rxns back to ice
Transform 1-2 uL of cooled rxns (assembled DNA) into 20-25 uL of competent bacteria (e.g. Stbl3s, especially when cloning vectors with repetitive sequences like lentiviral plasmids)
Thaw competent cells on ice for 5-10 min.
Usually we freeze 50-100 uL aliquots of homebrewed, chemically competent cells
Pipette gently to re-suspend/mix thawed cells then aliquot 20 uL of cells into pre-chilled strip tubes
Add cooled Gibson rxn (1-2 uL) to cells and swirl pipette tip to mix. Incubate cells+DNA for 5-30 min on ice.
Heat-shock cells 42C for 30s then return to ice for 2-3 minutes
Dilute cells with 200 uL of room temp. LB then either incubate 37C for 1 hr or plate immediately (if vector include ampR/carbR gene)
Spread 50-100 uL of diluted, transformed cells on agar plates w/ appropriate antibiotic
Incubate plates O/N at 37C (upside down)
The next day, count # of colonies (transformants) per plate.
Positive control: 23 colonies
v3 library assembly: 21 colonies
Great, both Gibson rxns appears to have worked. In afternoon, I picked 6 colonies from the sample plate and inoculated in 4 mL of LB+carb to grow O/N at 37C, 220 rpm
Next day, mini-prepped all 6 grown cultures eluting the purified plasmid in 25 uL of EB per sample
500-1000 ng / uL per plasmid
Submitted plasmids for long read sequencing by Plasmidsaurus
Next day, results were in. 4 of 6 transformants assembled correctly.
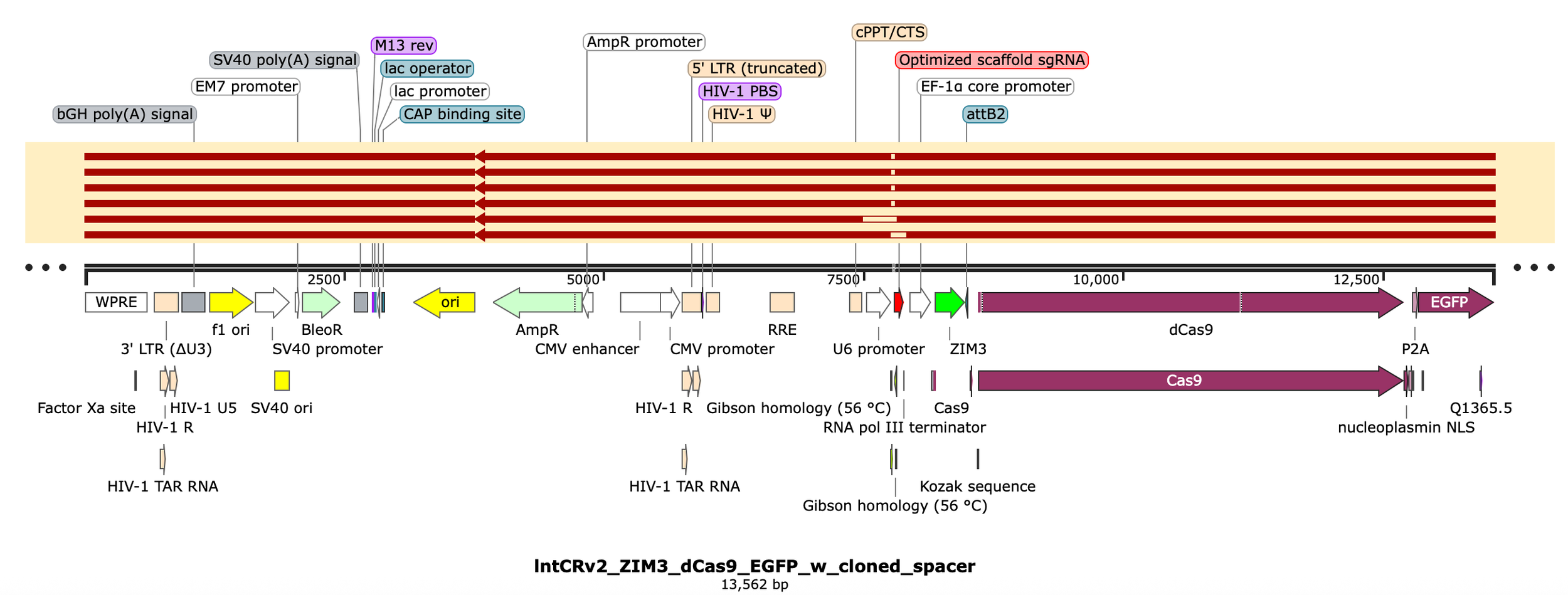
Zooming in to the sgRNA region…

Okay so 4 constructs appear to be clean assemblies while the other 2 are missing both the insert and few hundred bases of flanking sequence. This likely happened from T5 exo chewing back a much longer portion of the 5’ ends of the backbone followed by microhomology mediated annealing of the two exposed strands and repair (either within the rxn or later on in cells).
Ignore the random sequence tags (“Gibson homology, 56C” and “universal flanking sequence, SR lentiCRISPRv2“) as they were created by someone else and not used in this approach
And herein lies our problem w/ Gibson assembly for library cloning. The inclusion of T5 exonuclease (and Taq ligase) is likely to promote aberrant assemblies by creating long exposed ssDNA ends. These exposed ends (of variable length b/c we don’t have precise control over T5 exo activity) at 50C will, at some efficiency, find a sequence to anneal to whether that’s the intended insert or the other end of the backbone itself and re-circularize/repair leading to a significant background of plasmid byproducts (here, 33%) in addition to correct assemblies.
While this byproduct rate often doesn’t matter for “one-off” cloning experiments where you just need a single correct transformant, it becomes a huge problem when you’re (1) using a vector w/ a lot of repetitive sequence like a lentiviral backbone by raising the frequency of mis-assemblies considerably and (2) trying to expand a mixed population of correct assemblies as done in library cloning. While you may start with a proper assembly efficiency >70% in the completed Gibson rxn, once these products are transformed into bacteria and propogated O/N, the final pool of plasmids you purify can be dominated by aberrant assemblies if they preferentially expand in the population b/c of attributes like a shorter size and/or retainment of only essential components (e.g. the origin of replication and ampR cassette). This is exactly what I observed when I took the remainder of Gibson rxn that produced the 6 transformants sequenced above^ and transformed it into electrocompetent cells which I then expanded in a large culture O/N and maxi-prepped the next day to generate the “final” cloned CRISPR library.
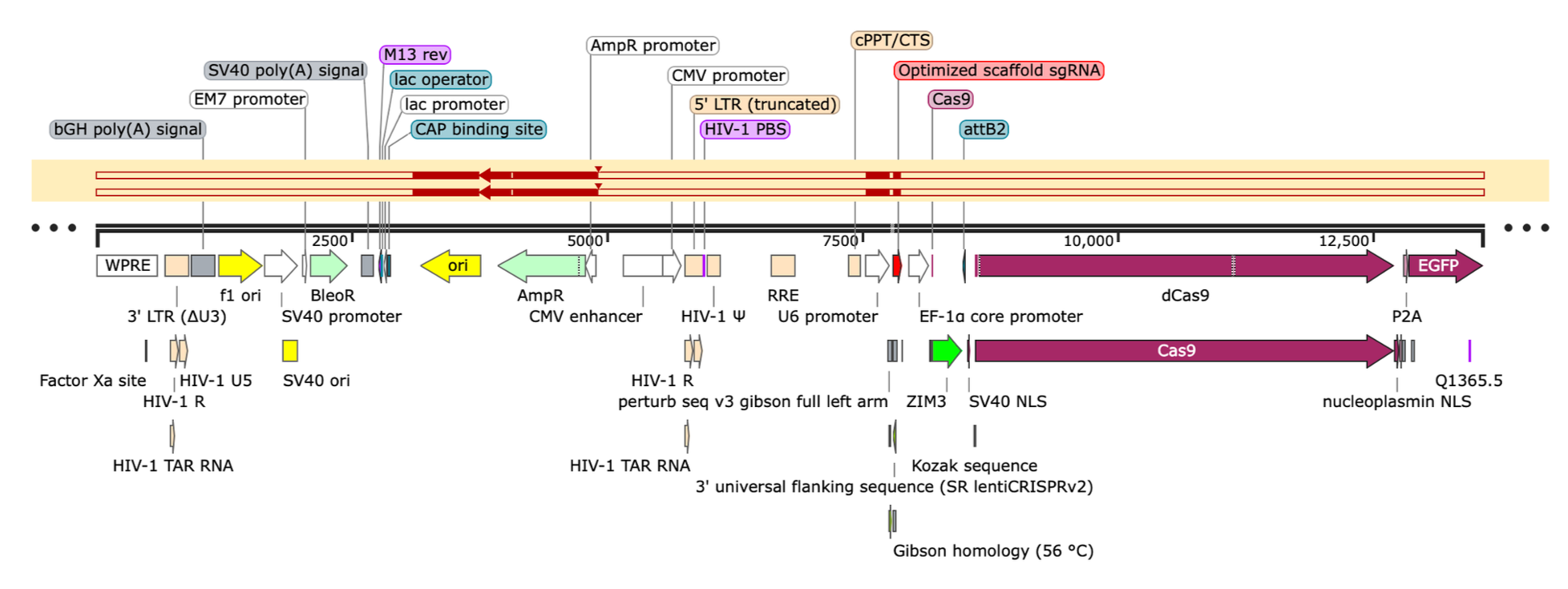
Sequencing replicates of the expanded plasmid library cloned by Gibson, you see above^ that a single plasmid has outcompeted the rest and come to dominate the pool. This plasmid is an aberrant assembly in which the backbone rearranged (presumably during cloning) to only include the essential pieces (ori & ampR) allowing it to preferentially propagate inside the bacteria.
Repeating the Gibson rxns a few more times under varied conditions (insert to backbone ratio, incubation time), I repeatedly observed aberrant assemblies interspersed amongst successful ones when plating individual transformants and consistently obtained a pool of recombined plasmid similar to above^ whenever I then tried to expand these assembled products to obtain the final CRISPR library (which is necessary to have sufficient material for downstream experiments).
Therefore, I conclude that Gibson assembly is not ideal for cloning CRISPR libraries in this manner (inserting small spacers adjacent to a highly structured scaffold sequence in a repetitive, lentiviral plasmid) and turn to our alternative method: circular polymerase extension cloning (CPEC) for which we can conveniently use the exact same insert & backbone fragments.
CPEC rxns
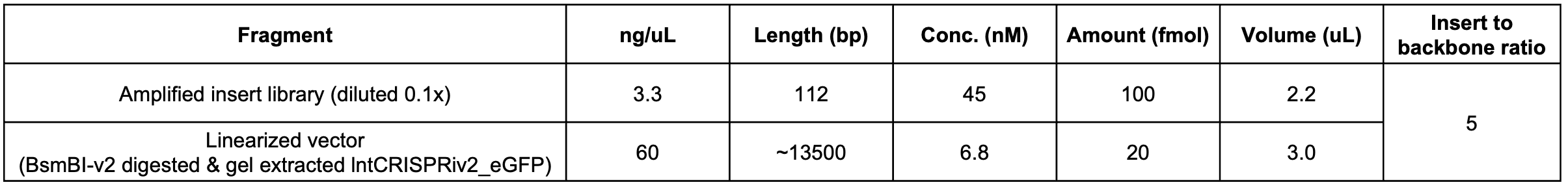
1. Thaw reagents (amplified insert library, linearized backbone, PCR 2x MM), vortex and spin down briefly, and place on ice
I’m using NEB Ultra II 2x MM here to have more control over the annealing/extension temp since Phusion Plus 2x MM recommends their universal annealing temp of 60C which is lower than I want
Theoretically, you don’t even need to amplify the insert library you can just input the synthesized pool received from TWIST directly into the CPEC rxn (although I haven’t tested this, there may some PCR inhibitors from synthesis)
2. Prepare 4 rxns (25 uL / rxn), as indicated in table below, in PCR strip tubes.
Ideal insert-to-backbone ratio is 5:1 or 10:1 (although you can go up to 100:1). Here, I used ~7:1

3. Vortex briefly to mix components then spin down tubes to collect liquid at bottom
4. Thermocycle for the indicated # of cycles (note that this is linear PCR amplification not exponential b/c we only have 1 “bridge” primer, the insert)
98C for 30s
[98C for 10s then 72C for 7 min] * 1-5 cycles … assuming a polymerase speed of 30s/kb * 13.5kb vector ~= 7 min extension
72C for 2 min
4C hold
5. Once completed, store rxns on ice or at -20C until next step
6. Purify CPEC rxn product by SpriSelect
Add 75 uL dH2O / rxn —> Now 100 uL / tube
Add 60 uL SpriSelect beads / rxn (0.6x Spri to purify dsDNA fragments > 600bp)
Vortex to mix. Incubate RT for 5 min then apply to magnet for 2-3 min
Wash 2x with 75% EtOH
Dry beads on magnet then elute DNA with 12 uL dH2O / rxn
Transfer 11 uL of eluate / rxn to new strip tubes
Measure by Nanodrop
CPEC rxn A aka "1+") 12 ng/uL (2.1/2.1) ----*12uL---> 144 ng (vs. ~130ng input)
CPEC rxn B aka "1-") 11 ng/uL (2.2/2.1) ----*12uL---> 132 ng (vs. ~120 ng input)
CPEC rxn C aka "5+") 17 ng/uL (2.0/2.0) ---*12uL---> 204 ng (vs. ~130 ng input)
CPEC rxn D aka "5-") 15 ng/uL (1.9/1.8) ---*12uL----> 180 ng (vs. ~120 ng input)
7. Based on DNA yield alone, it's unclear if CPEC is working although it’s a good sign we recovered more DNA from the 5-cycle rxns than the 1-cycle rxn
8. Proceed to test transformations (2 uL of purifed CPEC rxns / 23 uL Stbl3 competent cells)
Thaw 1 vial (100 uL) of Stbl3s on ice for 12-15 min then divide evenly across 4 strip tubes ---> 4x23 uL
Add 2 uL of purified CPEC rxn per tube
Pipette gently to mix and incubate on ice for 25 min
Heat shock 42C for 40s then return to ice for 2-3 min
Add 200 uL of room temp. LB / sample then plate 55 uL / rxn on LB+carb plates using Rattler beads (i.e. 1/4 of total cells per sample)
Incubate O/N at 37C in bacterial incubator
9. Next day, count colonies
A, 1+) 3 colonies
B, 1-) 0 colonies
C, 5+) 21 colonies
D, 5-) 0 colonies
10. CPEC appears to be working! To further validate, pick 3 colonies per successful plate (“1+” and “5+” rxns) and inoculate in separate tubes containing 4 mL LB+carb
11. Culture overnight 37C, 240 rpm
12. Next day, miniprep grown cultures and submit purified plasmids for sequencing (Plasmidsaurus, high concentration plasmid sequencing)
All 6 plasmid preparations were eluted in 25 uL per sample and measured ~2000 ng/uL by Nanodrop
13. Next day, results are in. All 6 samples are successful assemblies!
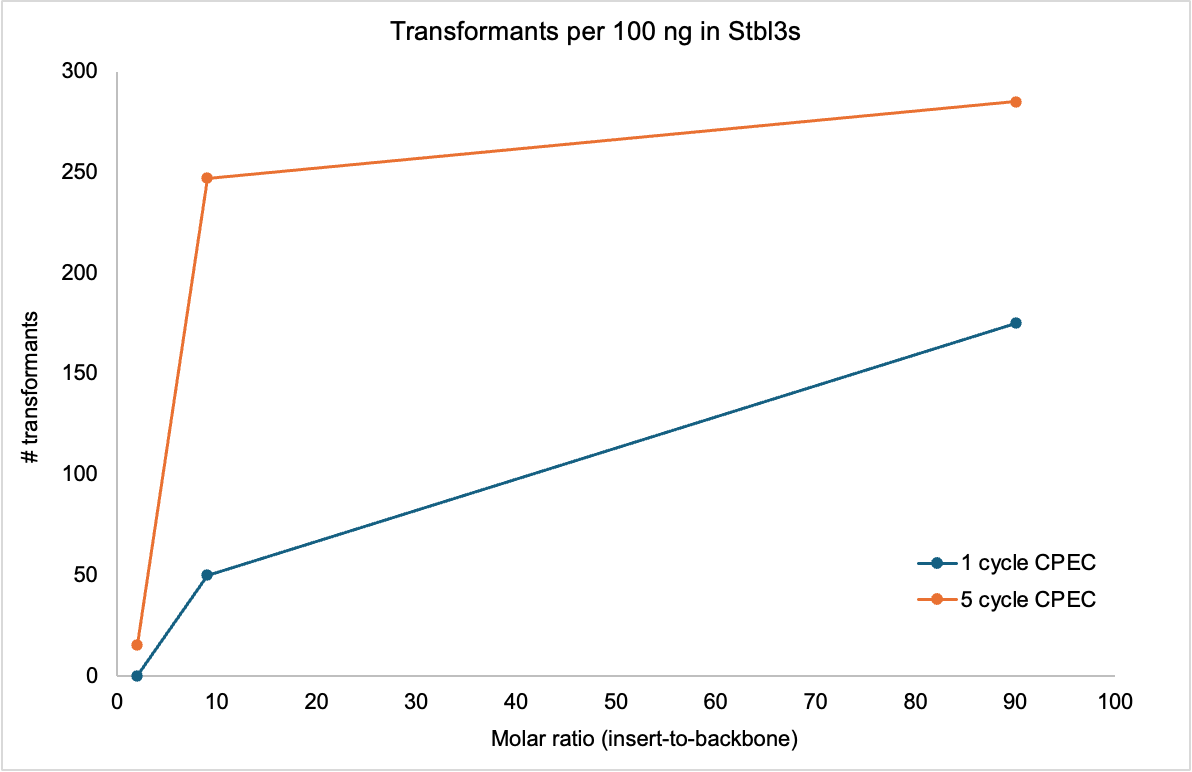
I tested the CPEC rxns a few ways and noticed these general trends. 5 (or more) cycles is advantageous but efficiency plateaus at an insert-to-backbone ratio of 10:1. Observed efficiencies are w/ Stbl3s (chemically competent) so you can expect ~100x more transformants per 100 ng (of purified CPEC product) using electrocompetent cells
Beautiful, now that we know CPEC is working let’s transform the rxn product into
electrocompetent Endura cells for library expansion
Stbl Transformation Efficiency:
One Shot Stbl3 cells > 1 x 10^8 cfu/µg (I use these for quick, heat-shock transformations)
Electromax Stbl4 cells: >5 x 10^9 cfu/μg
Endura Transformation Efficiency:
Chemically Competent Cells: > 1 x 10^7 cfu/µg
Electrocompetent Cells: > 1 x 10^10 cfu/µg (I use these for high efficiency/large-scale transformations)
14. Transform 5 uL of purified CPEC rxn product (from “5+” rxn C) into 50 uL of electrocompetent cells (1 vial of “Endura DUOs”)
I’m going to pass on using the “1+” rxn product although this condition did work
Applied ~90 ng of purified assembly product in total (can push this if you want and apply 200-400 ng of purified DNA to 50 uL of Enduras, however, keep total volume of DNA <=10% of total volume of cells)
Details of this electroporation step are included in “Cloning a CRISPR library”
Briefly, I followed Endura’s protocol exactly, keeping cells on ice throughout. Pulse in 0.1cm (1 mm) electrocuvette, 10 uF, 600 ohms, 1800V (pulse time should be ~5 ms)
15. Immediately after electroporation, dilute cells in 2 mL of recovery medium (warmed to at least room temp., ideally 37C) and recover in bacterial culture tube for 1 hr at 37C, 220 rpm
16. After 1hr recovery, dilute 10 uL of cell culture into 990 uL of recovery media (i.e. dilute 0.01x) and spread 100 uL of this dilution (after mixing) on LB+carb plate. Incubate 37C O/N
17. Dilute the remaining recovered cells (1.99 mL) into 110 mL of LB+carb in 1L baffled bottom shaker flask w/ vented top. Incubate culture 30-32C, 180rpm for 18-24 hours
The lower temp helps the library expand more evenly, highly recommend if you’ve got the time
18. Next day, count colonies on plate
100 uL of 0.01x dilution) 20 colonies ——> * (1000uL/100uL)*(2000uL/10uL) = 40,000 transformants total … this isn’t a ton but good enough for now (more CPEC cycles & more DNA added to Enduras should help increase)
40,000 transformants / 330 constructs = 120x coverage (not great, not terrible)
19. Purify plasmid library from large culture of transformed cells that expanded O/N
I like to split the culture into 2x50 mL aliquots (~10% of volume lost during 24h culture at 30C) pouring directly into 50 mL conical tubes
Process replicate samples in parallel w/ Zymo midiprep kit (makes it easy to keep centrifuge steps balanced), following the recommended protocol exactly including extra endotoxin removal step
Pelleting cells (3500xg, 10 min) from 50 mL aliquots of grown culture, I obtained large pellets weighing ~0.45g each (“wet weight”). Note: this is the max input per sample for the Zymo midiprep kit.
Eluted final plasmid in 200 uL ZymoPure EB / sample
Pool replicates for a total of ~330 uL of purified plasmid (some vol. lost during elution and filtering) & measure by nanodrop
v4 CRISPRi library) 540 ng/uL (1.9/2.3)
But did it (expansion) work??? Let’s check first by gel then long-read sequencing
Let’s do some old school DNA fingerprinting of the final plasmid pool (“CRISPRi library”) using EcoRI since there's a single cutsite in the final construct and we have EcoRI-HF in lab
88 uL dH2O
10 uL rCutSmart (thawed quickly at 42C and vortexed)
5 uL of v4 CRISPRi library (540 ng/uL)
2 uL of EcoRI-HF
Briefly vortex and spin down
Incubate 37C for 1 hr (no shaking)
Heat-kill enzyme 65C for 5-10 min at 1500 rpm
Purify digested DNA by 0.65x SpriSelect (washing 2x with 75% EtOH prepared yesterday)
Elute DNA in 50 uL dH2O, transferring 45 uL of eluate to new tube
Measure by Nanodrop
EcoRI digested v4 library) 43 ng/uL (1.9/2.3) ---*50uL----> 2150 ng (80% recovery)
Dilute purified digested library 1:20 (10 uL in 190 uL dH2O) and run on 1% agarose (EZ)gel for 10 min in triplicate lanes, next to the undigested CRISPRi library and 1kb Plus ladders
Undigested v4 library dilution) 540*(2.5/500) = 2.7 ng/uL *20uL = 54 ng loaded / lane (in lanes #1-3)
EcoRI digested v4 library dilution) 43*(10/200) = 2.15 ng/uL *20 uL = 43 ng loaded / lane (in lanes #4-6)
Expected size of linearized plasmids: 13.5kb
Next, submit an aliquot of plasmid pool to Plasmidsaurus for long-read ONT sequencing
Reminder that the returned consensus sequence is an averaging of all (aligned) reads meaning the spacer region will be incorrect / contain an extra, artifactual G (from experience) but the rest of the plasmid should be constant & match our reference sequence
Results returned next day (actually it was delayed in shipping and returned two days after submitting the AUDACITY)
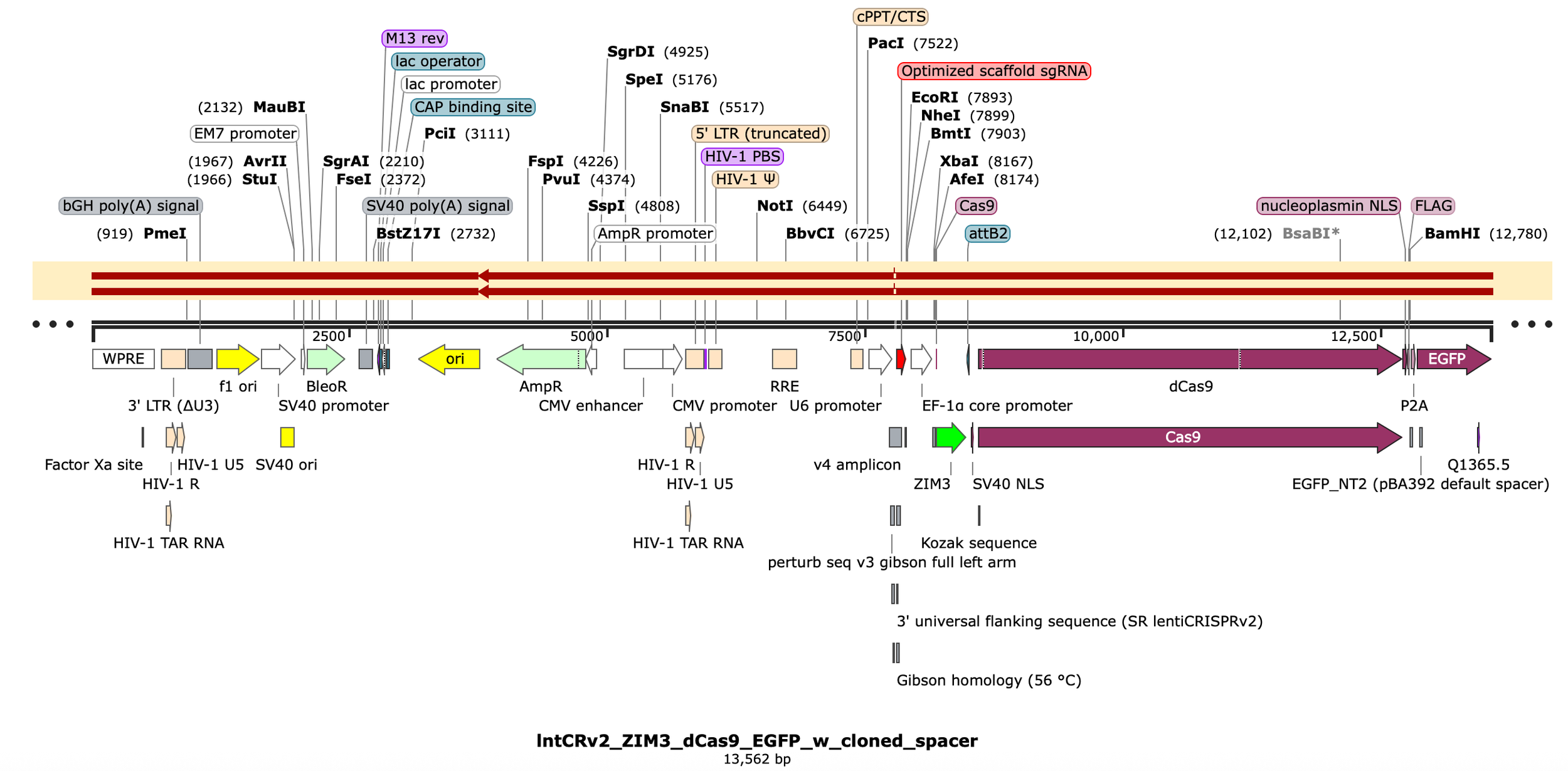
Consensus sequence for the pool is looking good - matching everything except the spacer region as expected
I submitted technical replicates of the final library to be certain since it will be used for an expensive perturb-seq experiment
What about the distribution of the raw reads in terms of total length? This is a good measure of library purity
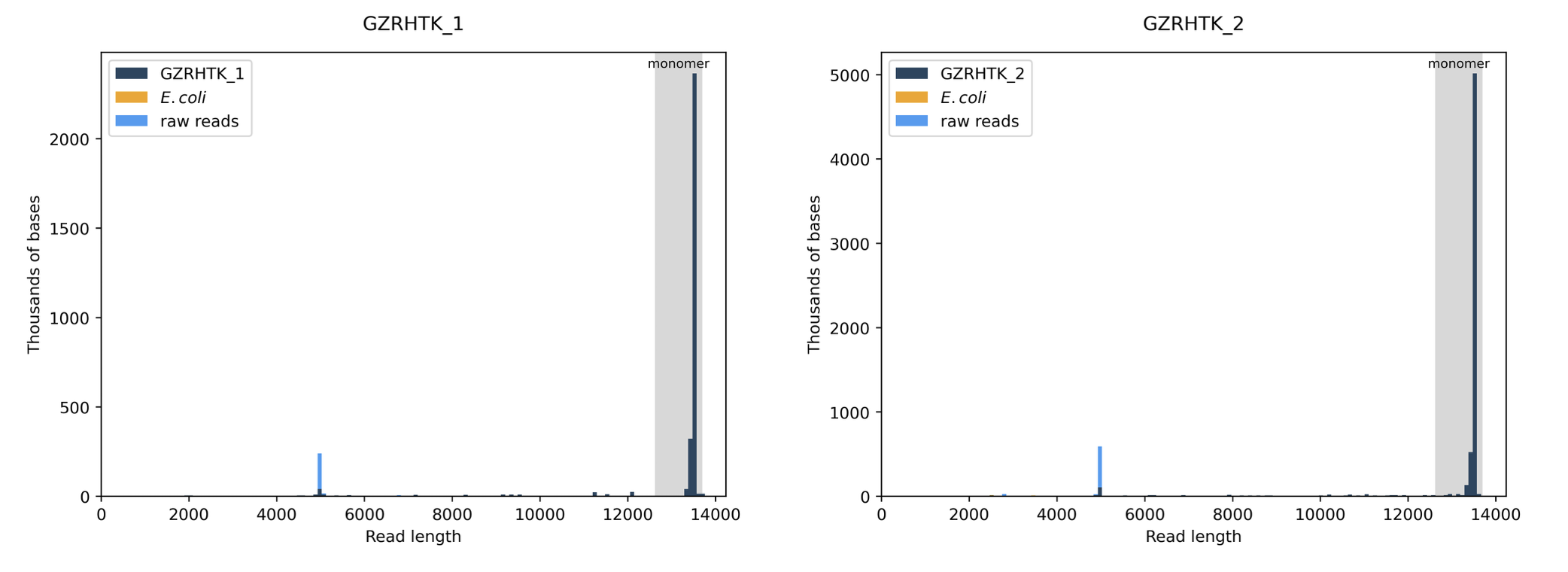
Thus far, our CRISPRi library cloned by CPEC appears to be cleanly assembled.
The final check? Library (spacer) diversity
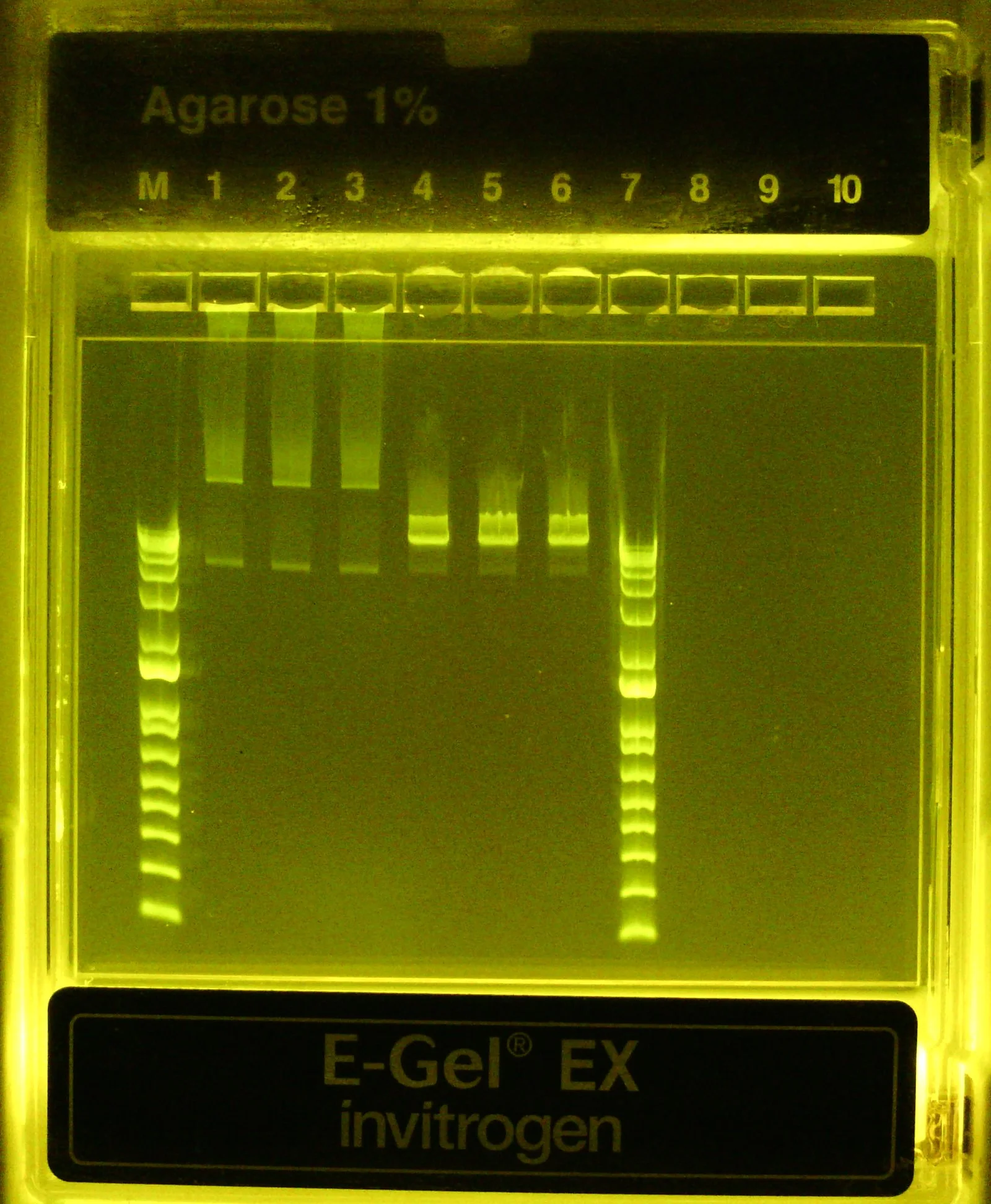
Okay cool, looks like once linearized the library is the expected ~13.5kb size (top of ladder is 15kb). I don't love that not all the DNA was converted (see faint band below primary one) after a 1hr incubation at 37C but perhaps it's hard for the RE to access the target site when the plasmid is supercoiled. At least we can confidently say MOST of the plasmid pool appears to be the correct construct. Before digesting, I interpret the two bands in the library to be relaxed/nicked plasmid (top band) and supercoiled plasmid (bottom band)
Okay pretty clean, most reads are the expected 13.5kb length… I don’t love this small population of “raw reads” (light blue) at 5 kb but I also don’t quite know how to interpret.. why aren’t they assigned to my sample? Or is it because they didn’t match the consensus sequence so they’re unassigned? Let’s check w/ Plasmidsaurus:
“Dark blue indicates that the raw reads align to the consensus/assembly sequence, orange maps to the E. coli genome, and light blue is unmapped, indicating either sequencing noise, a genome other than E. coli, or a lower abundance plasmid species that doesn't generate a consensus.” -
https://plasmidsaurus.com/blog/how-to-interpret-plasmid-read-length-histograms
Hmmm okay just what I thought. I’m not too worried about this even if it is another plasmid species in the sample as it’s lowly abundant even after expansion and unlikely to successfully package into lentivirus when we proceed to transfect the library into HEK293T cells but it may be worth looking into regardless to get a better sense of what it is and how it came to be (TO DO)
The final check
Let’s prepare Illumina libraries for short read sequencing the spacer region of assembled plasmids in the final pool to get a sense for how uniform the distribution of different spacers was maintained throughout the cloning/expansion process. We’ll do this the same exact way as described in the “Cloning a CRISPR library” protocol so I’ll only share critical details here.
In truth, our incredible undergraduate student Yanna did this part entirely on her own. Scroll down to see sequence details.
PCR1: 8x50 uL rxns
Use 10 ng of v4 library input as template per rxn (8th rxn = negative control)
oAEL052+Truseq F primer (100 uM): 5’ - ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCTTGTGGAAAGGACGAAA - 3’
oAEL053+Truseq R primer (100 uM): 5’ - TGGAGTTCAGACGTGTGCTCTTCCGATCTTGTTTCCAGCATAGCTCTT - 3’
Create PCR1 master mix (enough for 10 rxns)
25uL of Phusion Plus 2x MM * 10 = 250 uL
22 uL of dH2O * 10 = 220 uL
0.25 uL of forward primer (100 uM) * 10 = 2.5 uL
0.25 uL of reverse primer (100 uM) * 10 = 2.5 uL
Distribute 48 uL per strip tube (n=8)
Spike in 2 uL of 0.01x diluted v4 CRISPRi library (5.4 ng/uL) per tube (except for 8th, use dH2O)
Thermocycle:
98C, 30s
(98C 10s, 60s 10s, 72C 20s)* 12-14 cycles
72C, 2 min
4C, hold
Pool 7 positive rxns (8th is negative control) and purify by 0.6x reverse Spri selection (only keep supernatant) followed by 1.8x Spri selection (to enrich for small amplicons)
The first removes the plasmid template, the second purifies the small, ~140 bp amplicon. In reality, this is probably overkill, you can just do the 1.8X Spri to recover everything - it’s okay if some plasmid template carries through
Alternatively, can run a 1x Spri + 20% isopropanol to purify the small product → I recommend this! 400 uL pooled rxn + 400 uL Spri beads + 200 uL isopropanol works well to precipitate amplicon (then wash 2x w/ 85% EtOH)
Measure by Nanodrop & visualize by 2% agarose (EZ)gel
Pooled & purified PCR1 product) 22 ng/uL (1.7/1.8) ---*30uL----> 660 ng total
PCR2: 8x25 uL rxns (note: this is excessive, 1×50 uL rxn more than suffices if you do a calculation of molecular coverage)
Use 30-50 ng of purified PCR1 product as template per rxn (8th rxn = negative control)
PCR2 TruSeq forward indexing primer: 5’ - AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGAC - 3’
PCR2 TruSeq reverse indexing primer: 5’ - CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTC - 3’
Yanna chose a random set of indexing primers w/ structure outlined ^above (well H6 of pre-mixed PCR2 primer plate, i7 = ATGTCAGA, i5 = CTATCGCT)
Underlined regions are what’s annealing directly to the PCR1 product, the rest is being added on to adapt & index the fragments
Create PCR2 master mix (enough for 10 rxns)
12.5uL of Phusion Plus 2x MM * 10 = 125 uL
10 uL of dH2O * 10 = 100 uL
1.25 uL of 20 uM primer mix (10 uM per indexing primer) * 10 = 12.5 uL
Distribute 23 uL per strip tube (n=8)
Spike in 2 uL of pooled & purified PCR1 product (22 ng/uL) per tube (except for 8th, use dH2O)
44 ng template / rxn
Thermocycle:
98C, 30s
(98C 10s, 60s 10s, 72C 20s)* 6-8 cycles
72C, 2 min
4C, hold
Pooled 7 positive PCR2 rxns (8th was negative, no template control) for a total of ~170 uL
Purify/concentrate DNA by 1.5x SpriSelect, eluting in 30 uL dH2O. Repeat 1x.
Measure by Nanodrop & visualize by 2% agarose (EZ)gel
Pooled & purified PCR2 product: 25 ng/uL (1.9/2.3) —*25 uL—> 625 ng total
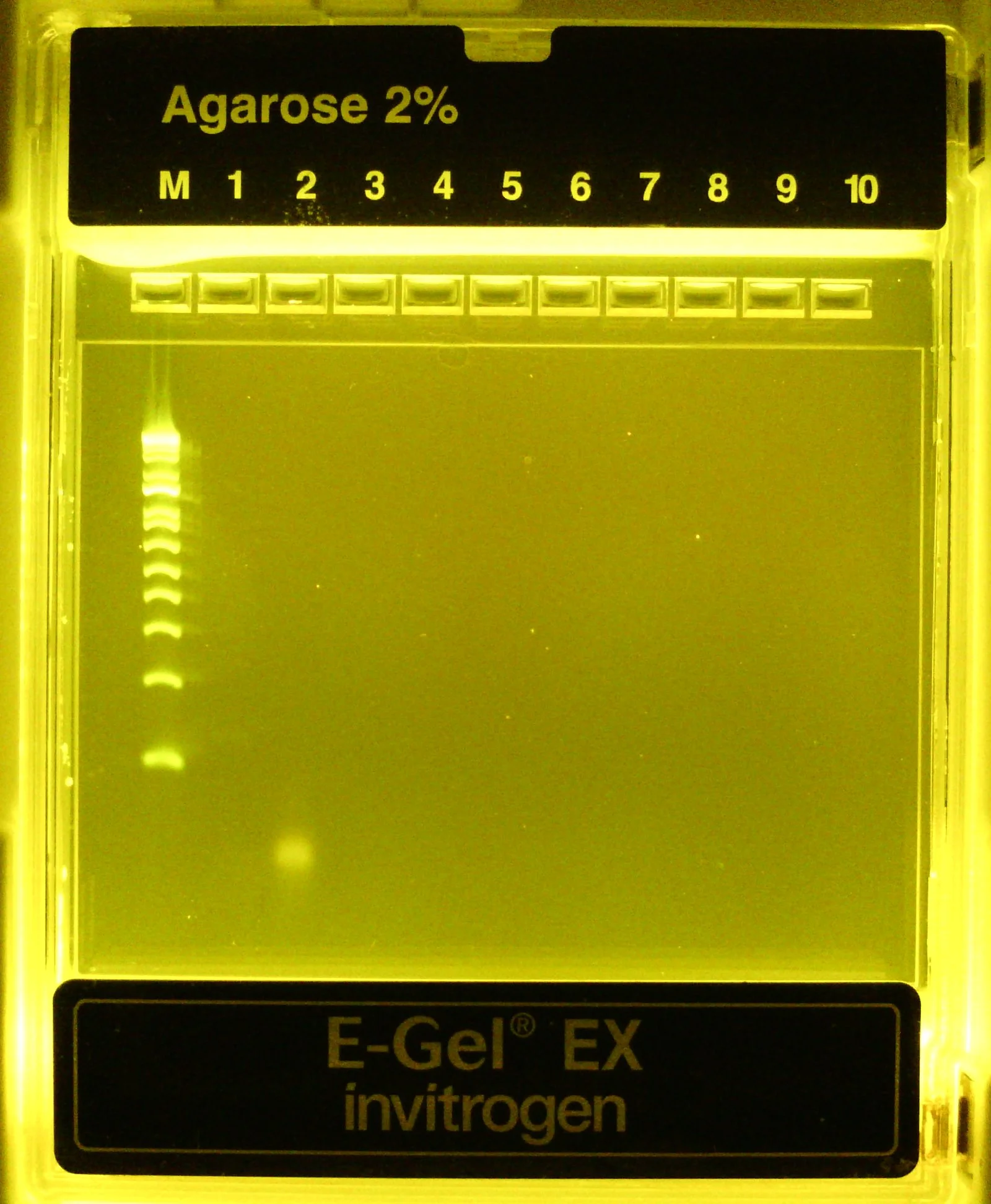
100 bp dsDNA ladder (M) followed by purified, 1:20 diluted PCR product (lane #1, it’s there I swear!) followed by 1:20 diluted negative control rxn product (lane #2). Very faint band at ~140bp visible for positive sample, strong primer dimers visible in control rxn
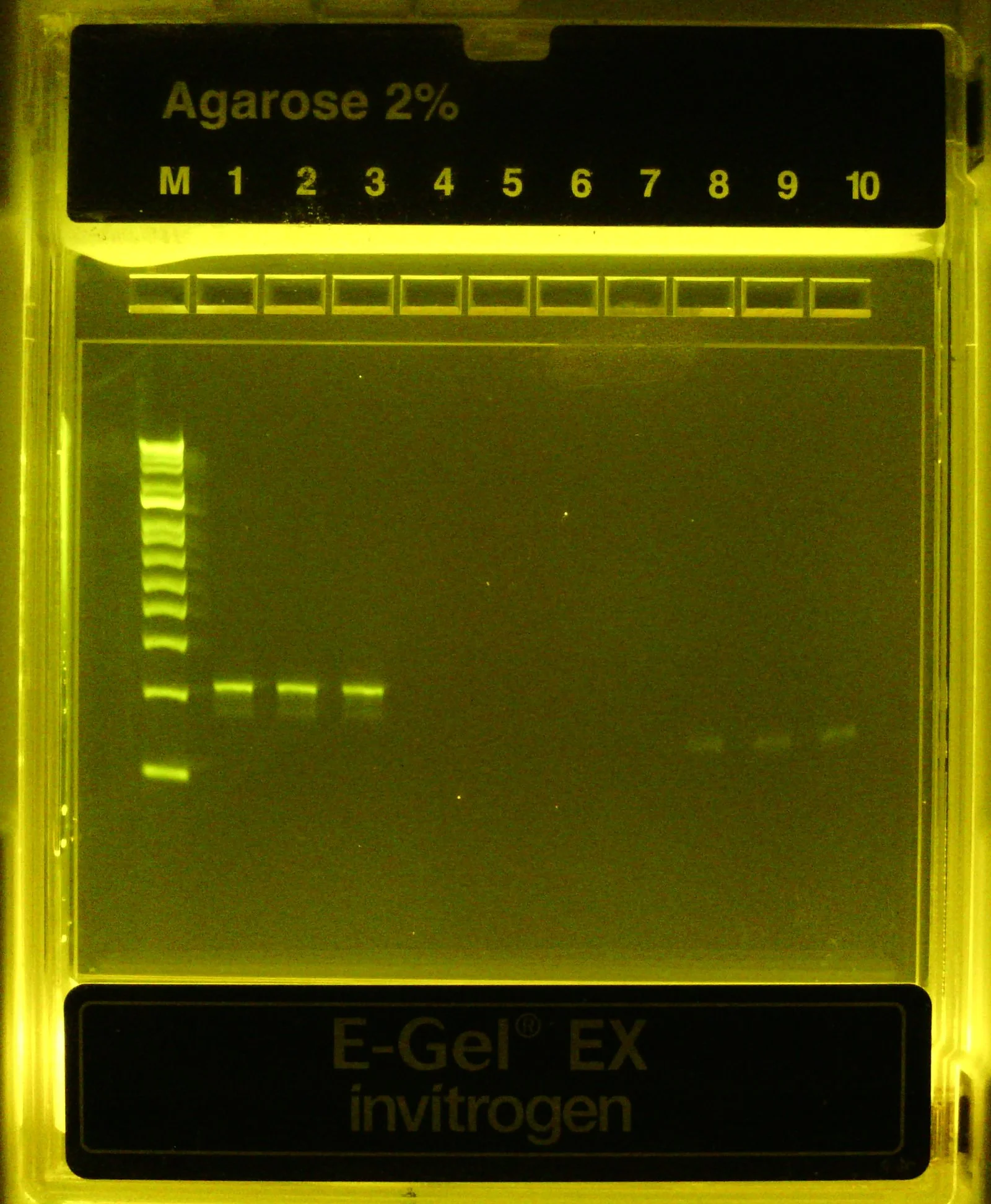
100 bp dsDNA ladder (M) followed by purified, 1:20 diluted PCR2 product (lanes #1-3) followed by 1:10 diluted PCR1 product (lanes #8-10). Clear band at ~200bp confirm successful PCR2 with adapter addition evident by the band’s shift upwards compared to PCR1 product (140bp)
PCR1 primers bind to target site on lntCRv2-derived plasmid containing cloned sgRNA spacer. Amplified region highlighted in blue (67bp)

PCR1 product (129 bp)

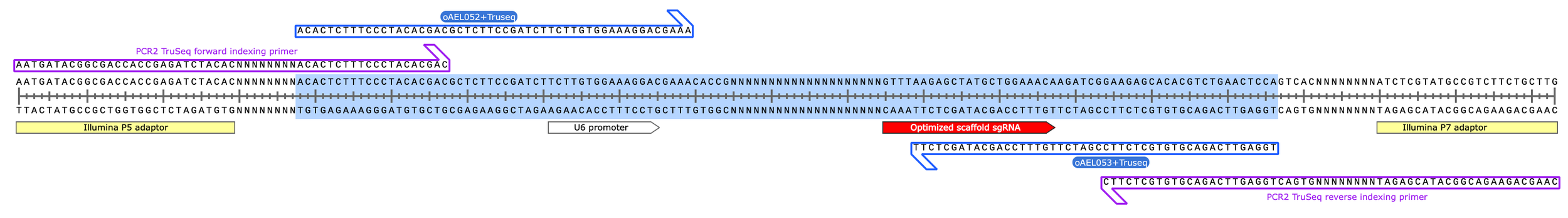
PCR2 product (203 bp)
(with PCR1 product highlighted in blue)
PCR2 product (203 bp)
(with Illumina’s sequencing primers indicated and original insert region highlighted in blue)
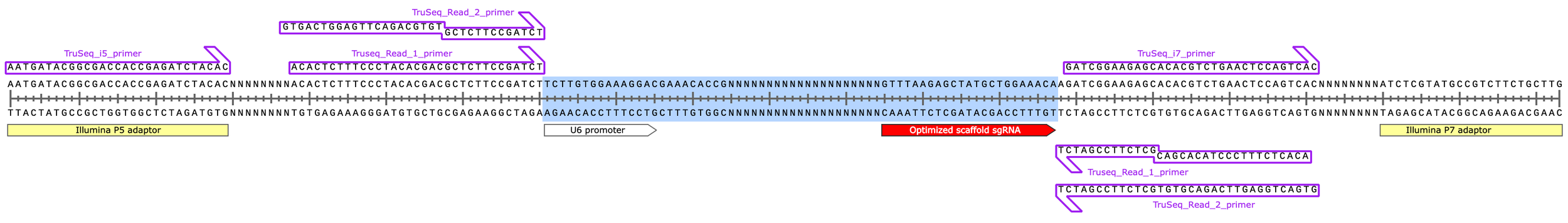
Note the secondary, “off-target” binding of the TruSeq Read 1 & 2 primers due to their identical 3’ ends (design feature of preparing sequencing libraries from blunt ended dsDNA fragments using Y-shaped adaptors instead of via PCR).
In reality, this off-target binding doesn’t occur because only one strand of the template DNA is present at a given time during sequencing
Next day, load validated PCR2 product (aka our “Illumina sequencing library“, different from the CRISPRi plasmid library we prepared it from) on MiSeq i100 for 25M, 100 cycle run
50 cycle x 8 cycle x 8 cycle x 50 cycle (paired-end) sequencing architecture (R1 / i7 / i5 / R2)
Since this is a single, small amplicon library, Yanna targeted a final loading concentration of 40 pM (instead of 100 pM) w/ 67% of DNA from prepared library and 33% from phi-X v3 control
Why do we do this? Small, single-peak amplicon libraries bind/amplify on the flow cell more efficiently than larger, bell-curve distributed libraries like phiX
See “High throughput sequencing w/ Illumina MiSeq” protocol for more details
Run completed successfully returning 29e6 total reads with 12% aligning to phi-X
You can see the effect I mention above in action, loading the divese/distributed phi-X library at a 33% molar equivalent resulted in 12% of total reads assigned to phi-X b/c amplicon libraries bind/sequence ~3x more efficiently
This means we have ~25e6 reads corresponding to the v4 CRISPRi library… perfectly played Yanna!
25e6 reads / 330 possible constructs ~= 75,000 reads per construct (i.e. sequenced incredibly deeply lol)
Final read analysis
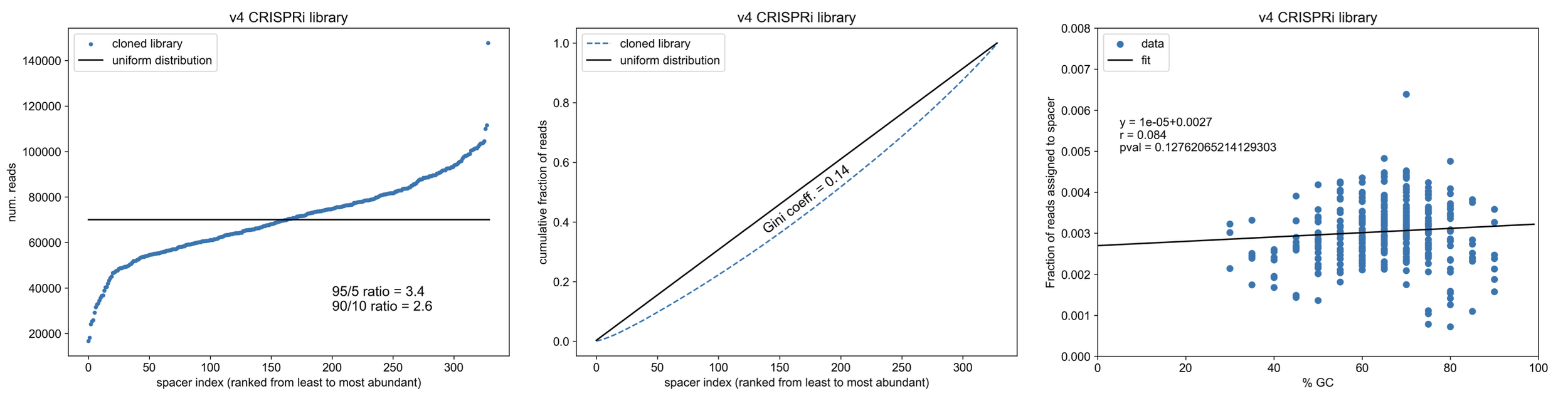
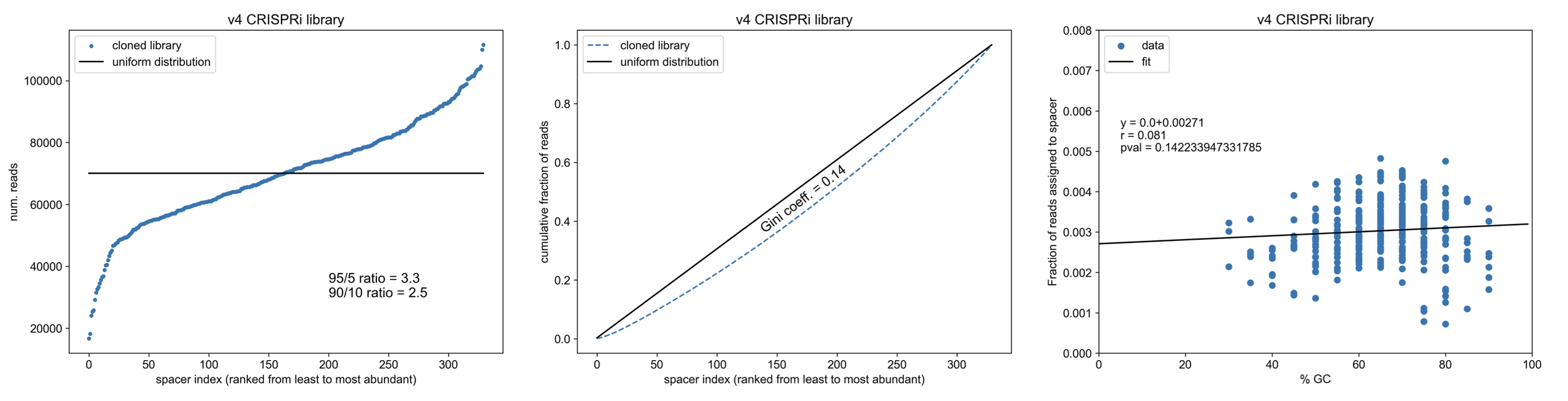
Gini coefficient = 0.14!
Please excuse the excessive number of digits reported in the p-values for the linear regression, I forgot round it and don’t want to remake the figures.
How does this compare to a similiarly sized (n=310 constructs) CRISPRko library cloned by restriction digest + ligation (as in my “Cloning a CRISPR library” protocol)?
Note the relationship b/w the GC content of cloned spacers and final library abundance - cloning such small fragments into the backbone via RE/ligation leads to biasing towards spacers that can stick together (not melt apart)
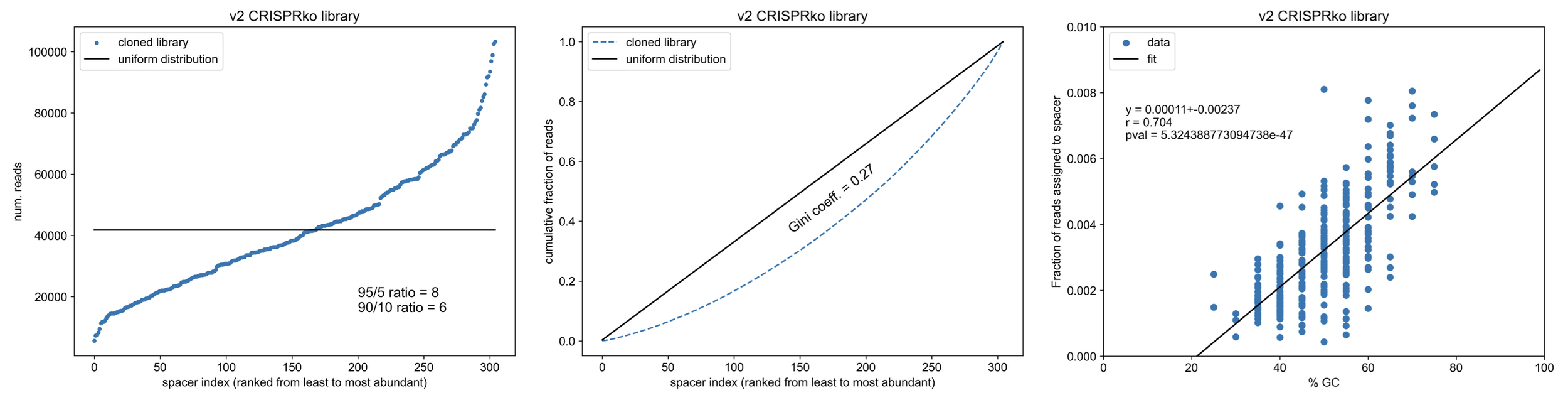
Wowwww, so CPEC knocks it out of the park compared to cloning by restriction digest & ligation. Not only is the method way easier & faster, CPEC doesn’t bias spacer integration based on GC content (except maybe at the extremes of %GC which is more likely a trend from oligo synthesis itself). Overall, our final CRISPR library is ~2x more uniform (less skewed).
Our final CRISPRi library is now fully validated and ready for experiments!
Summary
Let’s recap…
We set out to clone spacers into a lentiCRISPR backbone by annealing complementary, exposed ends instead of restriction digestion & ligation (which is also technically end annealing but you get the point but w/ much shorter overhangs). This meant we needed to include part of the U6 promoter and sgRNA scaffold sequence in the oligo designs
We immediately ran into an issue w/ this during the oligo library amplification PCR. Turns out, the sgRNA scaffold sequence is tricky and will easily fold on itself (as designed to..) to create secondary structures that are likely to (1) prime themselves and create aberrant byproducts during PCR when using a high-fidelity, proofreading polymerase and (2) block effective end annealing b/w fragments during Gibson assembly (we didn’t directly prove this second point but it’s very likely considering the low temp. used in Gibson assembly (50C) compared to the annealing temp (60C) used in the PCR where we ran into hairpinning issues).
We fixed the oligo design to avoid issues during the PCR, however, next ran into issues w/ Gibson assembly observing a high rate of aberrant assemblies (30-50% of transformants). This was likely to be caused by a combination of factors including (1) the exonuclease exposing long ends of the backbone that hybridized to each other and re-closed/re-combined the backbone (2) the exonuclease chewing through the entirety of our short inserts lowering the amount of substrate available for proper assembly and/or (3) hairpinning of the exposed scaffold sequence occluding fragment annealing as mentioned above and illustrated below.
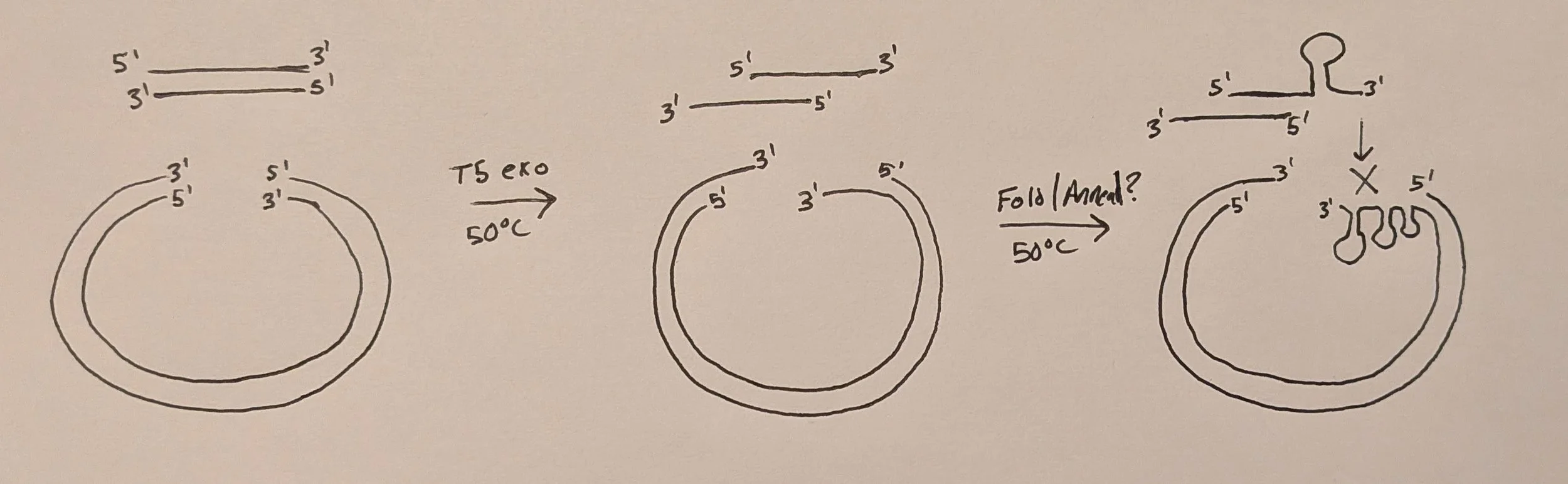
4. We confirmed this high byproduct rate during Gibson assembly was incompatible w/ library expansion via transformation into bacteria and then switched to CPEC
5. Using the same exact amplified oligo library and linearized backbone, we observed a 100% assembly efficiency (6/6 transformants) by CPEC
6. Transforming this CPEC product into electrocompetent bacteria, we successfully expanded the plasmid pool overnight to obtain a large, cleanly assembled library
7. Lastly, targeted sequencing revealed that library diversity was maintained (all spacers/sgRNA designs present) AND that construct abundance was highly uniform
Overall, I hope this was a helpful overview of an alternative method for CRISPR library cloning. I leave you with this comparison of the pros & cons of Gibson assembly vs. CPEC

Fun, extra fact…
Wondering what’s going on with this one overrepresented construct in the final v4 CRISPRi library? So was I, esp. b/c spacer GC content was normal
Turns out, I ordered two oligos w/ the same exact design, so of course it’s ~2x overrepresented!
How did this happen? Well I chose CRISPRi spacers from the hCRISPRi-v2.1 collection designed by Horlbeck et al (2016), selecting the top 3 spacers per gene EXCEPT in rare cases when the gene had multiple TSS annotations. In those cases, I chose the top 2 spacers per TSS.
In the one-off case of FOXP1, it had 4 unique TSS annotations so I chose the top 2 spacers for each TSS and 2 of the 8 spacers I ultimately selected happened to be the same b/c the annotated TSSs overlapped.
This double jeopardy was not accounted for when I assigned reads to spacers in the final analysis b/c I “collapsed” all the designs to unique spacers first. That’s actually where I should have noticed the discrepancy as I started w/ 330 designs but ended up w/ 329 unique spacers to which I then assigned/counted the reads (I just assumed it was a 0-based indexing thing)

Anyway, knowing what I know now here are the updated v4 CRISPRi library construct distribution figures (& stats) when you account for the doubly synthesized FOXP1 oligo
95/5 and 90/10 ratios improved slightly, as expected. Gini coefficient also improves from 0.137 to 0.135 although it’s not indicated b/c of rounding.
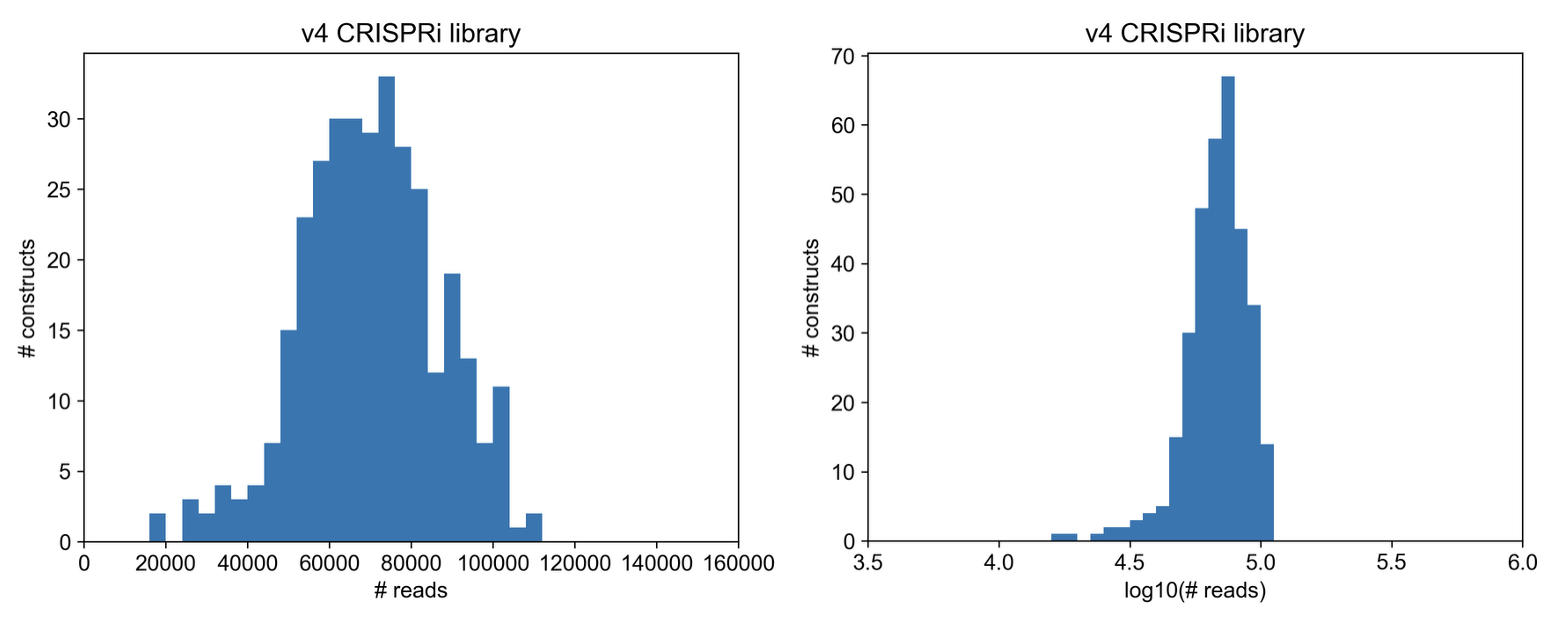
Here’s a direct view of the read count distribution (proxy for sgRNA distribution in cloned library)
Here are figures provided by TWIST (the oligo manufacturer) of an ideal (& unideal) library for comparison…
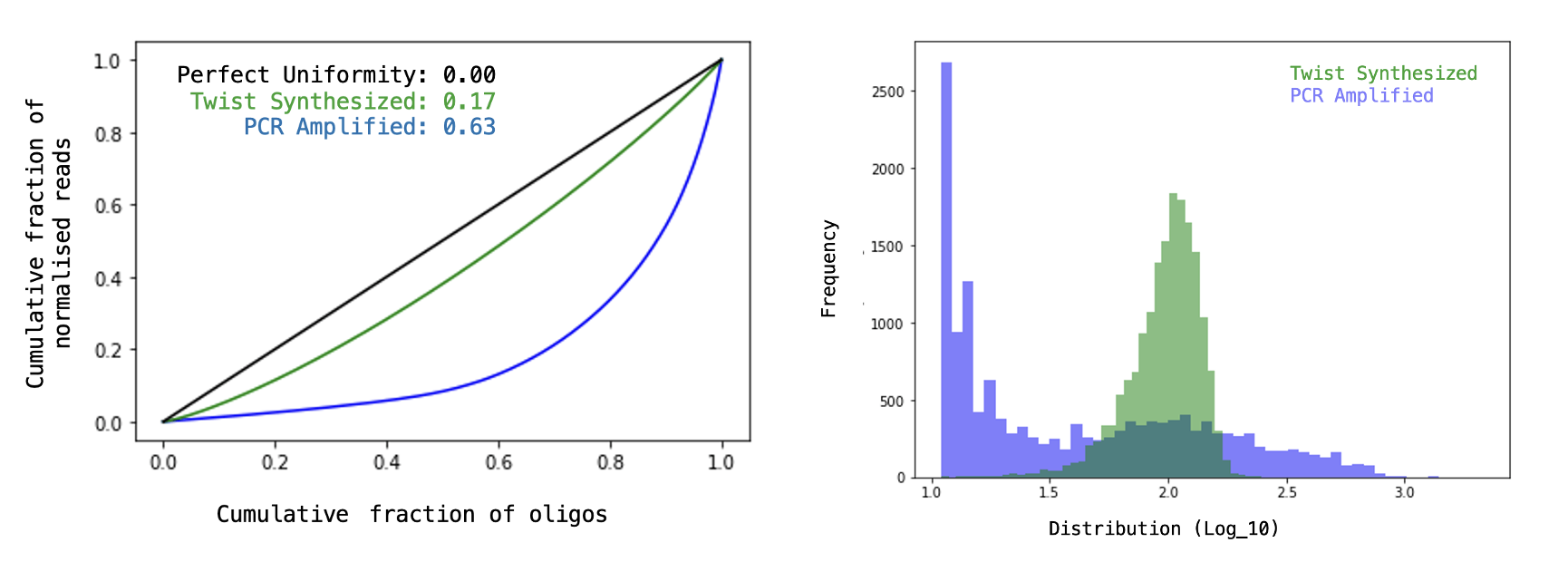
Full spread of constructs kept within an order of magnitude! That’s very good.
In other words, CPEC exceeded expectations
Follow-up
I helped a colleague clone another small CRISPRi library (n=366 sgRNAs, based on Repair-seq library that focuses on genes involved in DNA repair) and CPEC worked beautifully once again. This time, the transformations seemed to go even smoother as we got >200,000+ transformants per electroporation (50 uL Endura DUOs + 5 uL DNA @ 10-20 ng/uL)
90% of reads were assigned to a specific spacer/sgRNA based on perfect matching, the other 10% contained small errors. This error rate matches what we'd expect from synthesis, TWIST claims a success rate of 99.5% per base therefore (0.995)^20=90%
Interestingly, in each library, the most enriched sgRNA was ATR_-_142297649.23-P1P2 (5’-GGCCCGACGGAGCCGTGTGG-3’). This spacer is extremely GC rich (80%) and has some partial complementarity to the puroR gene included in our lenti vector so if there's any leaky expression in E. coli that induces a metabolic burden on the cells it could be alleviated by antisense binding of the sgRNA to the transcripts followed by endogenous RNA decay. I think, for this reason, it is one of the guides that is often cited as a “jackpotting” construct in CRISPR library cloning/expansion. While not a big deal here (ATR sgRNA is only 1.5x more abundant than any other sgRNA), people are developing other ways to propagate libraries to avoid this effect