Isolating white blood cells from leukapheresis product
So ya want to do some research in primary human cells, huh? Here are lessons learned from my trials & tribulations isolating primary human T cells and monocytes.
Before we begin, it’s important to confirm the cells you’re interested in are present in peripheral blood (i.e. what’s circulating in your blood vessels), otherwise you better collaborate directly with a research/clinical hospital with access to patients willing to donate other tissue. Blood is great because it’s minimally harmful / invasive to sample (i.e. relatively quick & easy to collect) as well as relatively easy to preserve and ship. That being said, I fully recognize giving blood isn’t a pleasant human experience, especially by leukapheresis which takes 2-4h, so I sincerely and deeply appreciate anyone who has voluntarily provided theirs for research and hope/expect you were duly compensated for it (typical rates are ~$100/hr).
What’s blood? Basically a cell-rich protein shake.
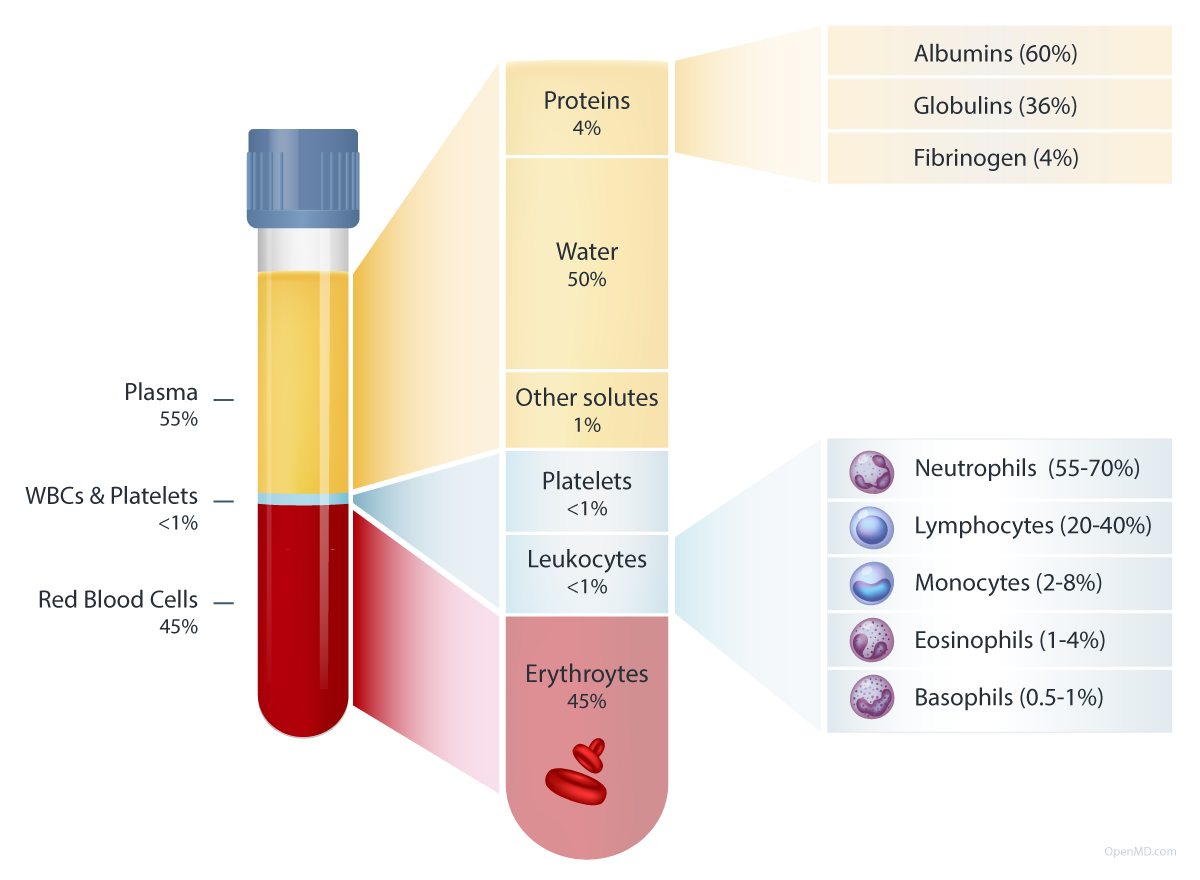
Great figure from OpenMD (https://openmd.com/guide/blood-components)
Details
Percentages in figure are volumetric, by mass blood is 91% water, 7% proteins, 2% other
Proteins at lower abundance in blood include antibodies, cytokines, and complement proteins.
Examples of “other solutes” include ions, nutrients, gases, hormones, and waste products
Platelets (aka “thrombocytes”) are not cells themselves but small sticky fragments of former cells called megakaryocytes. Their main purpose is to clot for wound healing / tissue repair.
Leukocytes = “white blood cells”. Their main purpose is defense (immunity).
Erythrocytes = “red blood cells”. Their main purpose is oxygen transport.
The avg. adult has ~5L of blood in circulation. Interestingly, the max amount you’re allowed to draw/donate at once, or total within any 8 week period, is 500 mL (i.e. 1/10th of your total)
5000 mL * ~1.055g/mL (blood density) = 5.275kg * (2.2lb/kg) = 11.6 lb i.e. 5-10% of your body weight is blood. Obviously weight can vary but I’m not sure if total blood scales or if everyone just makes do w/ roughly 5L… Update: it scales but nonlinearly (specifically, logarithmically or “with diminishing returns” for you business folk)
Where do blood cells come from?
They all trace back to hematopoietic stem cells (HSCs) in the bone marrow. The process of HSCs developing/differentiating/maturing into distinct blood cells is known as haematopoiesis. The final cell types listed above are considered “terminally differentiated”. In other words, they’ve finally settled into a fixed class/state in order to perform a specific function in the body.
How exactly do HSCs transition into all these different blood cell types? The working model of the human hematopoietic lineage tree is a bit of a mess. I hesitate to even show you the “classical model” b/c it’s been so heavily revised over the last 10-20 years with the rise of single cell technologies (as well as improved flow-cytometry & isotopic tracing techniques).
I like this visual summary (on the right) of our evolving understanding from https://doi.org/10.1016/j.cell.2020.08.030. Essentially, it’s a continuous cascade - early on HSC derivatives may start leaning towards one trajectory or the other based on signals they encounter but they remain susceptible & able to changing trajectories as they’re exposed to more signals.
This is pure conjecture but you can imagine it’s best for the system (human) if blood cells remain as pluripotent/flexible as possible for as long as possible in case any new informations arises (e.g. signals from an infection). With this reasoning, I’d say that true & total commitment takes place when a cell develops a feature that is crucial for the core function of the final lineage but physically incompatible with the core function of another cell type (e.g. RBCs commit relatively early when they eject their nucleus to maximize oxygen storage after which they can no longer jump into another trajectory)… otherwise why have different, specialized cell types at all?
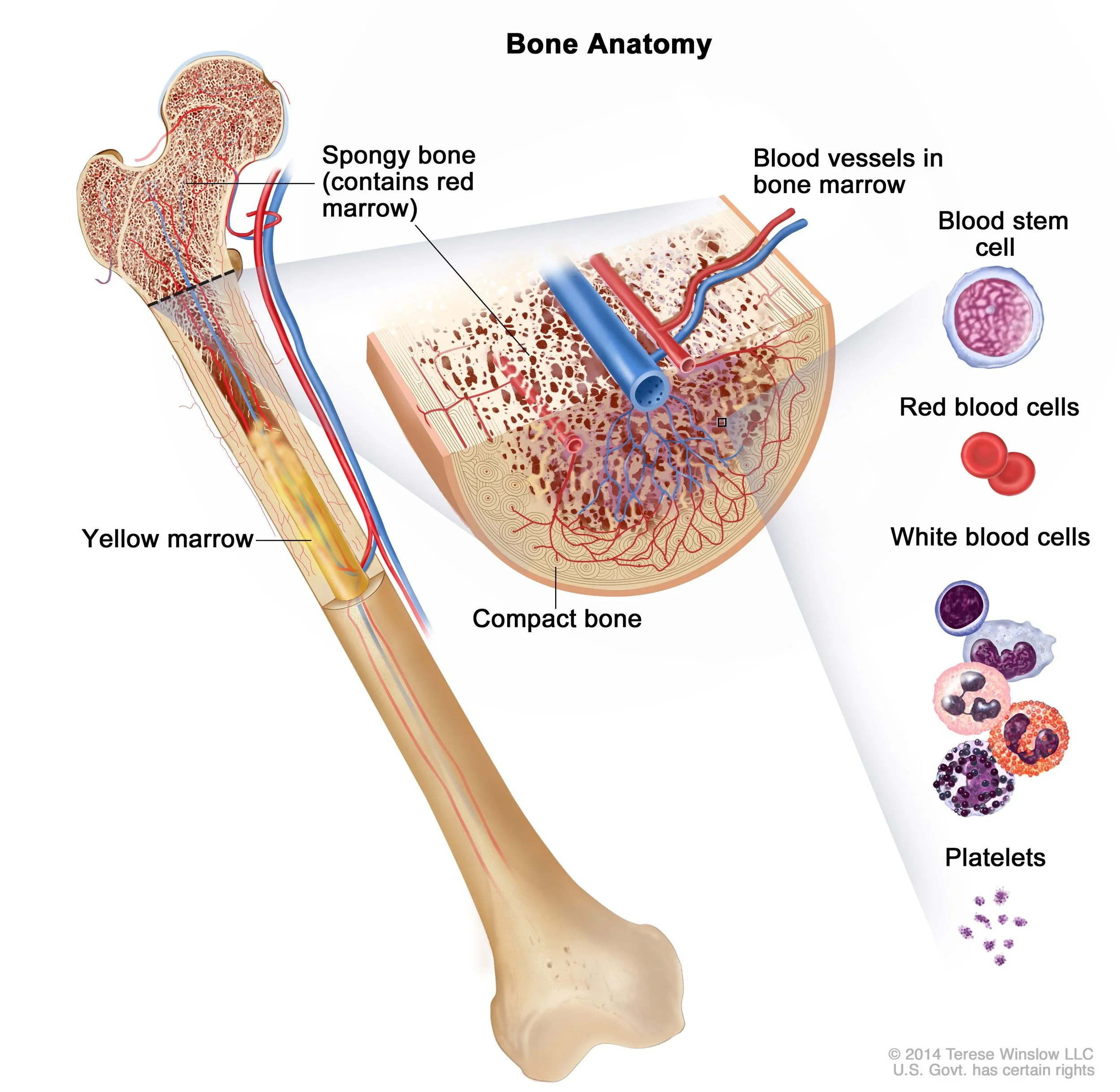
Your body generates >100 billion (1E11) new blood cells per day to maintain a steady state (!!!)
Also, you may notice macrophages (technically another white blood cell) are missing from figure on right. Most macrophages in your tissue were seeded there during early development and then just persist by dividing / self-renewing. If more macrophages are needed, they come from monocytes which are constantly circulating in your blood waiting for certain signals to pounce into action. Once these signals are received, monocytes quickly exit the bloodstream and enter tissue at the site of distress, then differentiate into cells with a morphology/role resembling macrophages AND/OR dendritic cells.
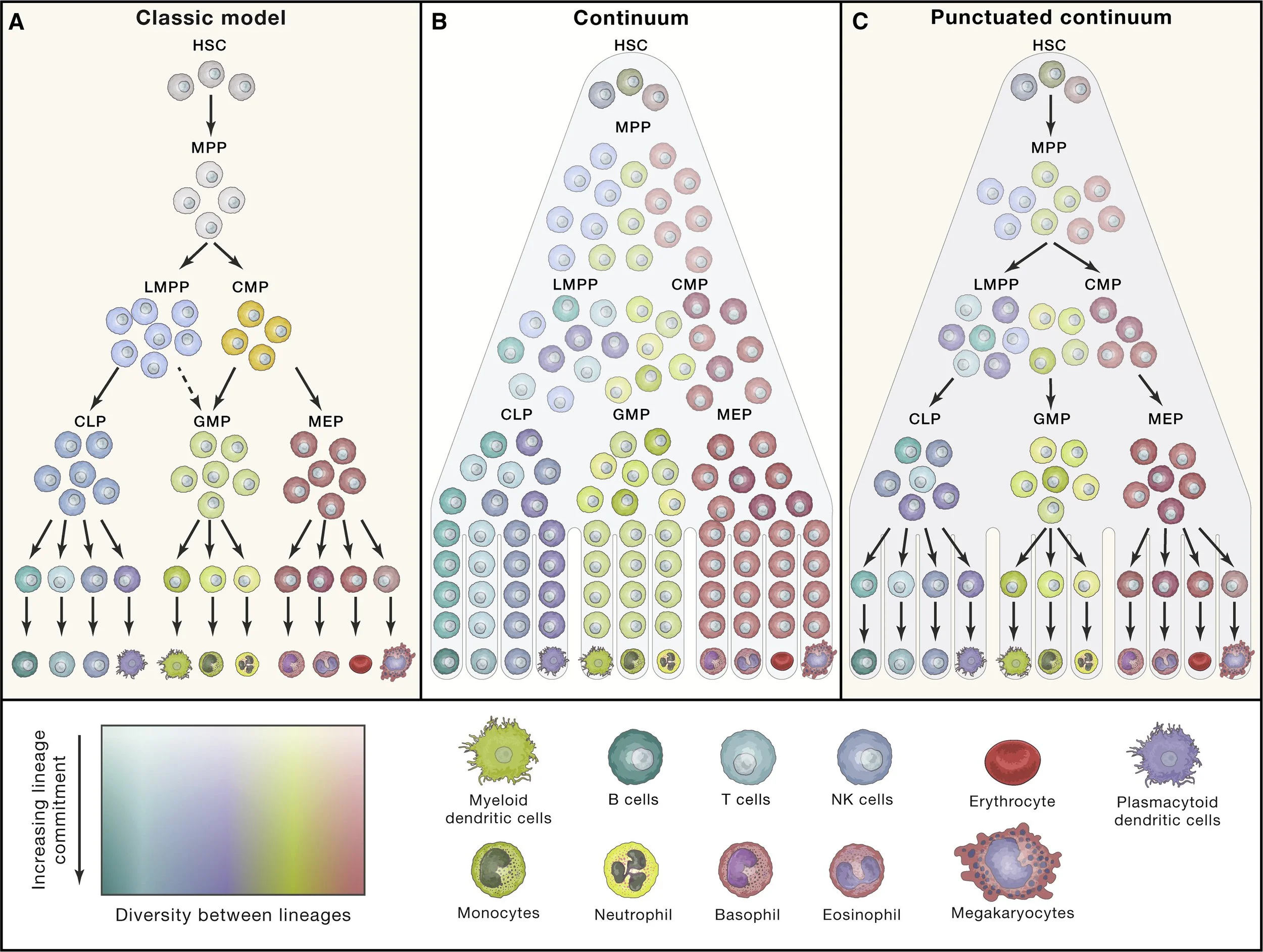
Liggett, L.A., and Sankaran, V.G. (2020). Unraveling hematopoiesis through the lens of genomics. Cell 182, 1384–1400
Generated w/ nano banana. No idea why it picked this style when prompted for a “diagram”, reminds me of patent illustrations.
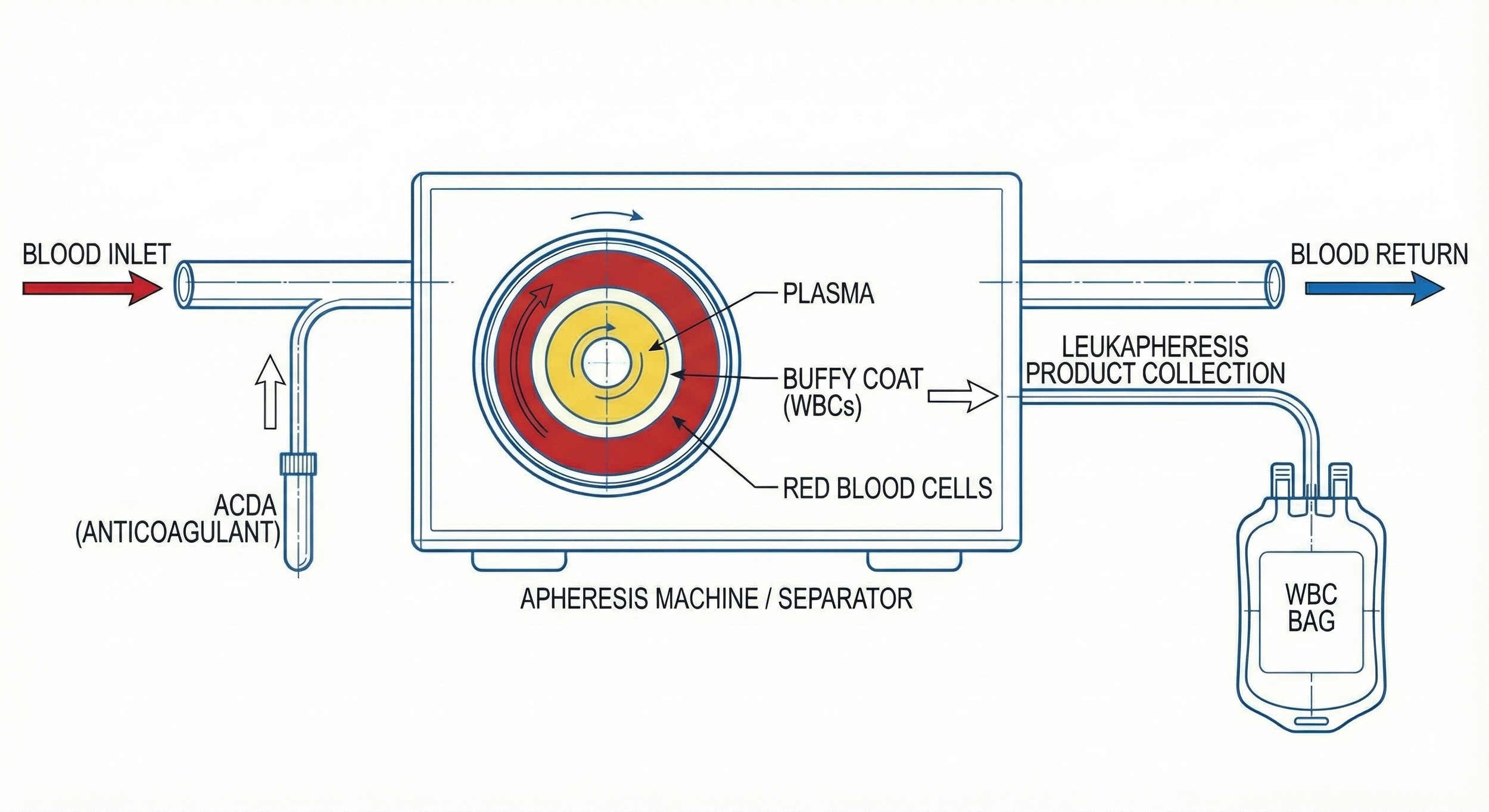
What’s a “Leukopak”? A sample of blood enriched in white blood cells by a process called leukapheresis.
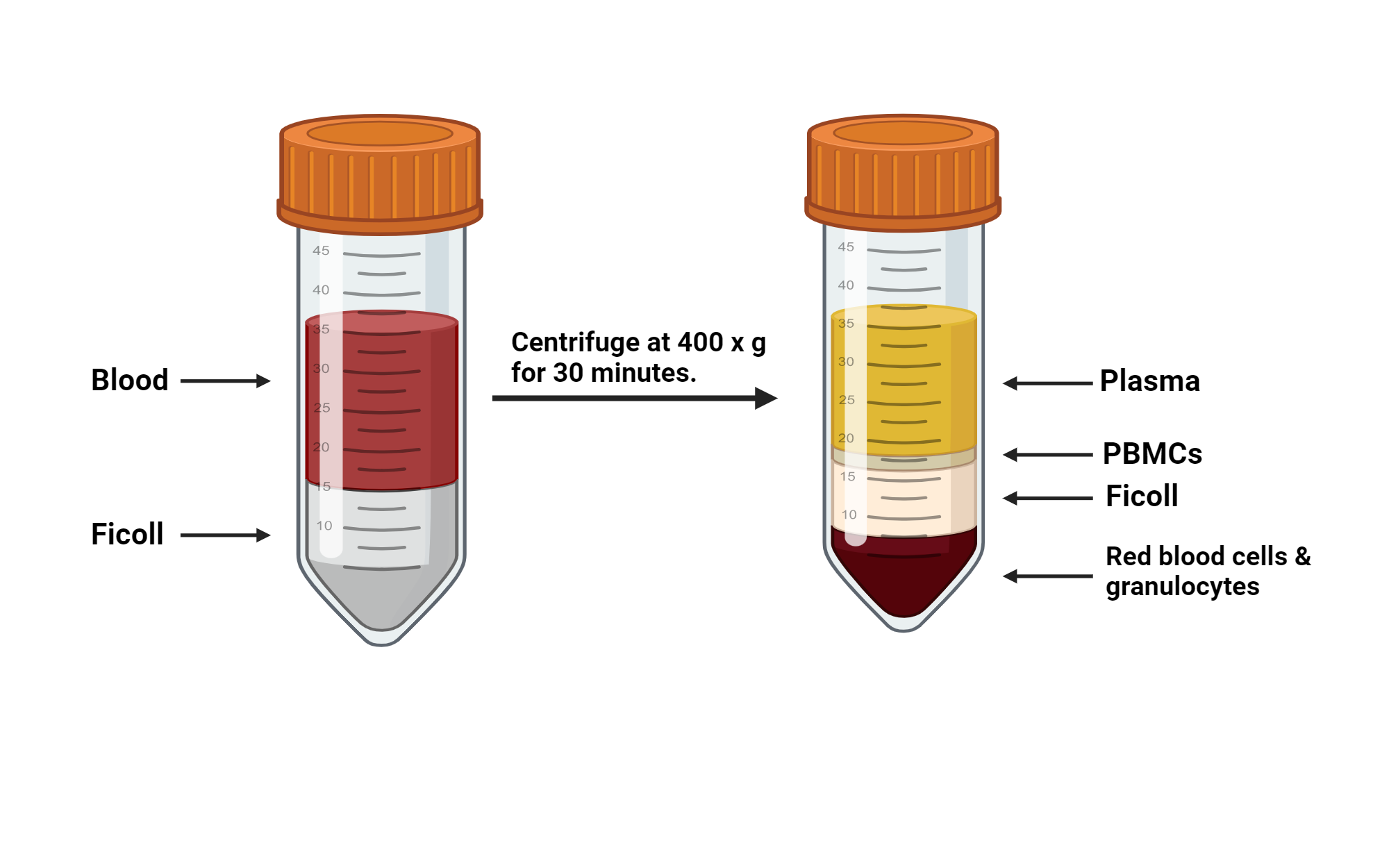
Most commonly, leukopaks are collected by continuous flow using an aphaeresis machine wherein blood is drawn from one arm, immediately mixed w/ anticoagulant to prevent clotting, and the passed into a centrifuge to separate blood components by density. The desired (mononuclear) white blood cells, which are more dense than plasma but less dense than red blood cells & granulocytes, are then separated into a collection bag while all that remains is returned to the donor through their other arm. This design minimizes wasted material while greatly enriching the cells of interest.
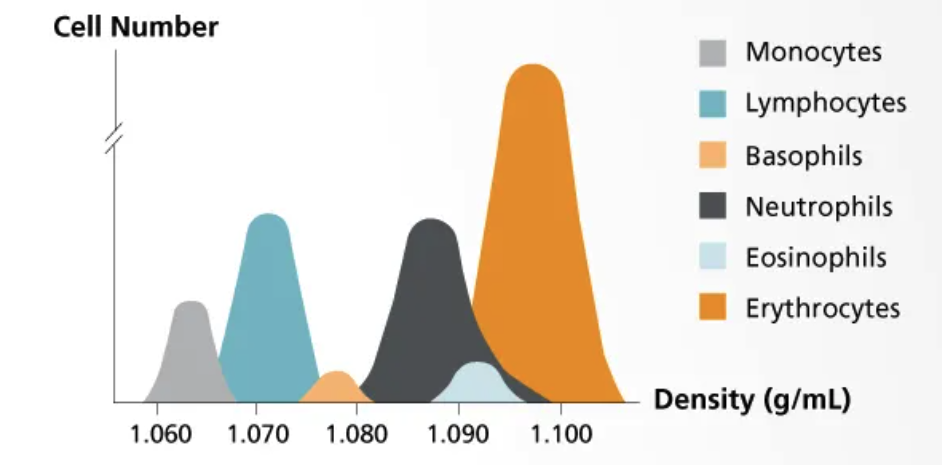
As you can see, the ideal density to separate monocytes & lymphocytes from the rest of the blood cells (RBCs, granulocytes) is ~1.08 g/mL
Lymphocytes+monocytes = cells in peripheral blood w/ single, non-lobed nucleus therefore they’re consider “peripheral blood mononuclear cells” or PBMCs
From STEMCELL (www.stemcell.com/cell-separation/methods)
Why not just use whole, peripheral blood?
You very well could but let’s run the numbers…
Conc. of red blood cells (RBCs) in peripheral blood = 5e9 cells/mL (damn!)
Conc. of platelets in peripheral blood = 3e8 cells/mL (~10x fewer than RBCs)
Conc. of white blood cells (WBCs) in peripheral blood = 7.5e6 cells/mL (~1000x fewer)
Notes
These #s can vary widely across patients (and their state of health), I’m using averages for healthy adults
Typically, these values are presented as cells / uL or cells / mm^3 (equivalent) in a medical context
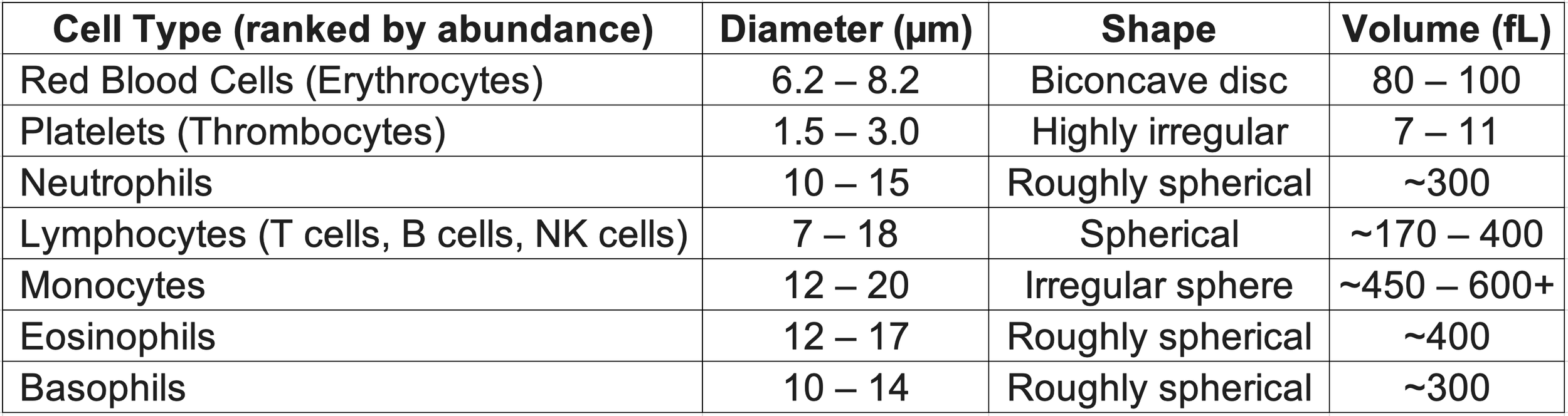
Setting RBCs as the standard, platelets are ~10x smaller while leukocytes are 2-5x larger by volume
For comparison, in lab we typically culture cells at a density of 0.1-1E6 cells/mL…
If a single donor were to provide 1 pint (473 mL) of blood, the max allowed, we would obtain…
5E9 cells/mL * 473 mL = 2.4e12 red blood cells
7.5E6 cells/mL * 473 mL = 3.5e9 white blood cells
These are huge numbers, no doubt. 2 trillion RBCs?? Pretty much incomprehensible. As many WBCs as half the humans on the planet??? Damn. You would think this would be enough cells to last years of research but, unfortunately, that’s not the case. First, multiply the total # of WBCs by the fraction you actually want (e.g. 30% of WBCs are T cells). Next, we’ll work with 1E5-1E6 cells per “sample” to get a robust average signal due to modern day detection limits of most instruments/assays. Multiply that by often 10s-100s of different samples (constituting different conditions/treatments & replicates) tested simultaneously in 6-, 12-, 24-, 48-, or 96-well tissue culture plates and you quickly run out of cells after only a few experiments.
*
*not cells but I wanted to include. You can argue RBCs aren’t either b/c they have no nucleus (in humans)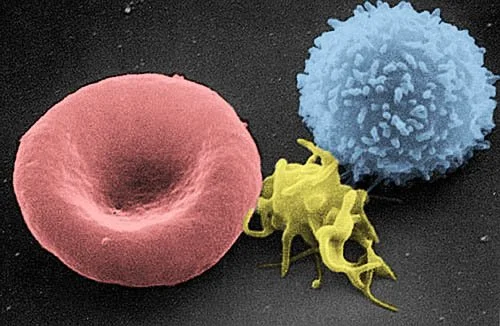
From left to right:
Red blood cell (RBC), platelet, and lymphocyte. Image from the Electron Microscopy Facility at The National Cancer Institute at Frederick (MD)
Okay, so what about a leukopak?
Typically, 150-300 mL of leukapheresis product can be collected in a single donation
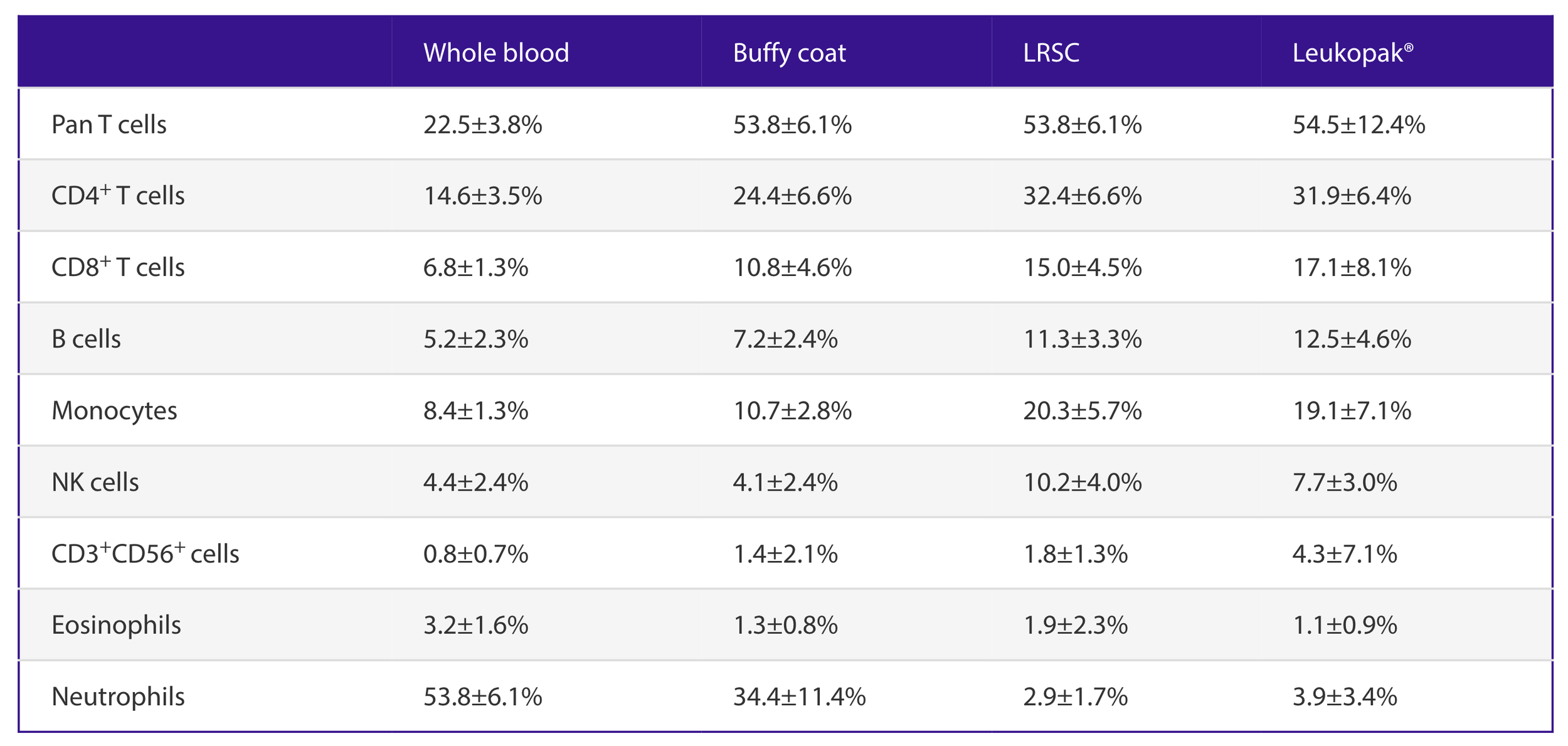
I really like this breakdown of cell composition in leukopaks from Miltenyi Biotec:
You can see leukapheresis enriches for mononuclear cells (T cells, B cells, monocytes) while depleting red blood cells and granulocytes (neutrophils, basophils, and eosinophils)
Total cell yield in a leukopak?
RBC concentration ~= 7e7 cells/mL ———*200 mL ——> ~1.4E10 total (>100x depletion)
WBC concentration ~= 6e7 cells/mL ———*200 mL——> ~1.2E10 total (>3x enrichment)
So not only do we obtain ~3x more total WBCs than from a collection of whole blood, it’s also significantly “cleaner” (depleted of red blood cells and neutrophils) and in less volume
Another moment of appreciation to any donors as leukapheresis withdraws ~30% of all white blood cells circulating in your body. Amazingly, they naturally replenish back to baseline within ~3 days.
You can actually physically see the difference in RBC & WBC content with leukopaks being lighter red / more whiteish yellow than whole blood
Okay enough background, how do we actually isolate/purify cells from a leukopak? Immunomagnetic separation!
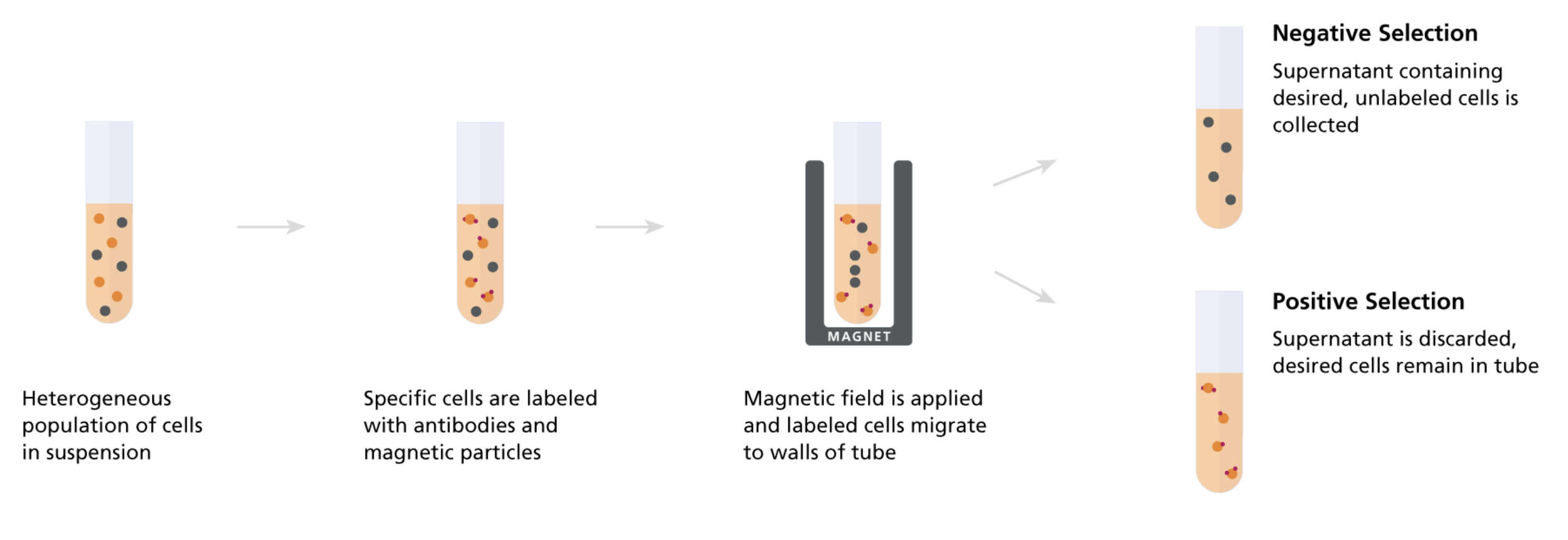
I use a combination of cell separation by density gradient (i.e. Ficoll-Paque or Lymphoprep separation) followed by STEMCELL’s (negative selection) immunomagnetic kits containing antibodies that bind to the surface of cells as well as the surface of ligand coated, paramagnetic beads in order to purify a specific primary human immune cell type from leukopaks.
Density separation isolates mononuclear WBCs
(aka "peripheral blood monouclear cells” or “PBMCs”)
From BioRender
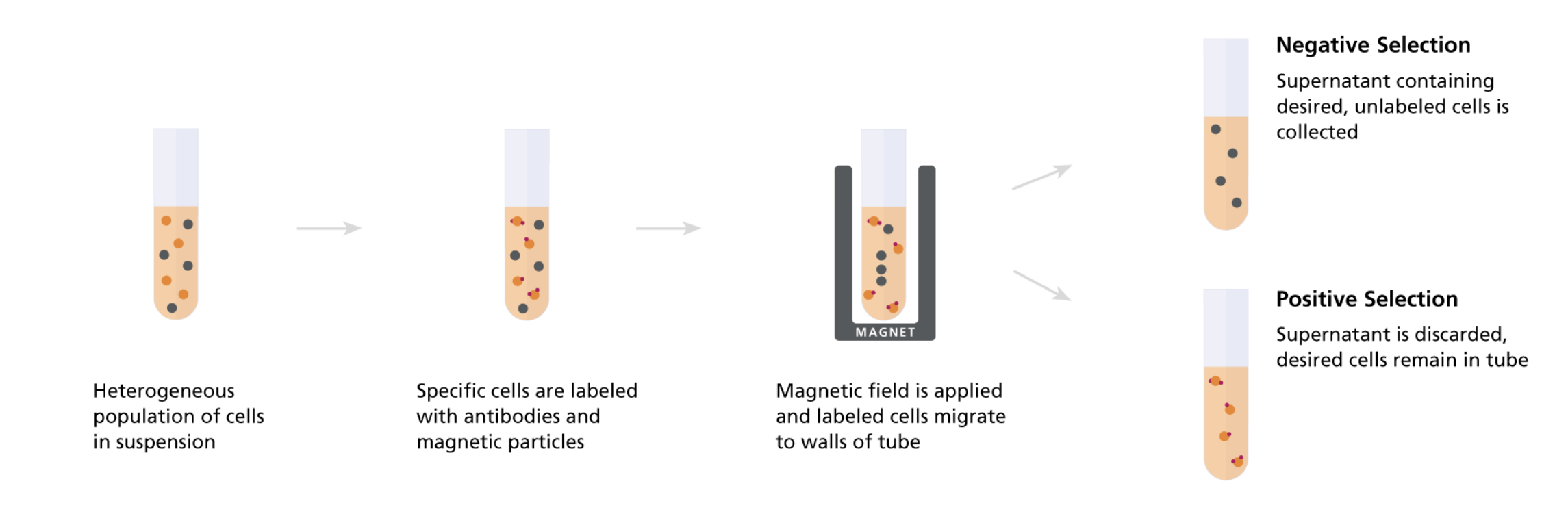
From STEMCELL (https://www.stemcell.com/cell-separation/magnetic-cell-isolation)
Immunomagnetic (negative) selection purifes target cell type by removing all others
I find that the initial separation-by-density step really helps clean up the sample by isolating PBMCs from undesired neutrophils & RBCs. Even though leukapheresis product is much “purer” than whole blood in terms of WBC content, it still has lots of unwanted material that risks clogging / disrupting the downstream immunomagnetic isolation step.
Why not just start from whole blood then if you’re going to density separate anyway? B/c leukopaks cut down significantly on the total volume you need to process, saving lots of time & energy in TC. However, you very well could follow the same protocol as below starting w/ whole peripheral blood (in some anticoagulant like ACDA).
Cost of a leukopak? Well usually I don’t require a full one. Instead, I’ll order a quarter leukopak (guaranteed to contain >=2E9 WBCs) from BioIVT for $1,700. I assume w/ economies of scale a full leukopak costs ~$5,000
This ends up being the perfect amount of starting material, as each STEMCELL kit can process up to 1E9 nucleated cells and I need to split my sample to isolate autologous CD8+ T cells and CD14+ monocytes in parallel
Protocol
Part 1: Stock up on required materials (mostly from STEMCELL to keep consistent w/ their kits) then order quarter leukopak and work w/ provider to schedule collection/delivery date
STEMCELL EasySep Human CD8+ T cell (negative selection) isolation kit (Cat. #17953): $1,000 …. for processing 1E9 total nucleated cells
STEMCELL EasySep Human (CD14+CD16-) monocyte (negative selection) isolation kit (Cat. #19359): $1,000 …. for processing 1E9 total nucleated cells
STEMCELL EasySep buffer (Cat. #20144): $120 / liter … or make yourself (dPBS + 2% FBS + 1 mM EDTA)
STEMCELL CryoStor CS10 (Cat. #07930): $525 / 100 mL … animal component-free, defined cryopreservation medium with 10% DMSO
STEMCELL Lymphoprep (Cat. #18061): $200 / 500 mL … Density gradient medium for the isolation of mononuclear cells
BioIVT Human LEUKOMAX quarter pak (Cat # HUMANLMX25-0001127): $1,700 … guaranteed >2E9 total “nucleated” (white blood) cells.
Why not just order the leukopak material from STEMCELL as well? You can, although I’ve found BioIVT’s to be cheaper & more consistent in quality (although based on a small sample size).
Importantly, order w/ ACDA as anticoagulant and for O/N shipping so that cells can be processed within 24-36h of collection. Can be shipped cold (4C) or at ambient temp., minor pros & cons to each
All together, I typically spend ~$4,000 for the consumables to isolate roughly 50-200 million CD8+ T cells and 100-200 million CD14+ monocytes (from 1/4 leukopak)
The quantities of buffer, cryopreservation medium, and density separation medium recommended above are enough for processing cells from 3-5 different donors (quarter leukopaks)
Note that you’ll also need to 1-time purchase a custom STEMCELL magnet for the separation step. I highly recommend an “Easy 50 EasySep magnet” purchased through Ebay ($500 vs $2000 from supplier)
$4,000 / 400 million primary human cells = $10 per million cells… as opposed to spending ~$90 / million cells if purchasing isolated cells directly (1 vial of 1E7 purified primary human immune cells costs ~$900)
These ~90% savings are a no brainer for an extra ~4 hrs of work IF AND ONLY IF you know you can reliably obtain such high yields from the EasySep kits. I’ve spent a lot of time troubleshooting the isolation process since I started out recovering only 10-20 million total primary cells from each EasySep kit (i.e. 5-10% of their advertised yield). I think I’ve now worked out all the critical details needed for consistently obtaining >75% yields.
Part 2: Divide, DNase treat, dilute, and density separate leukopak contents
0. Warm EasySep buffer to room temp (can do this overnight, just leave on bench). Thaw DNase I (5 U / uL, 100-200uL) and store on ice.
1. Open package containing leukopak, set aside documents
Usually documents include a shipping label/receipt, signed certificate of analysis, and info sheet with donor details & leukopak QC metrics
2. Quickly and carefully transport leukopak to biosafety cabinet
I’ll usually place the small bag in the base of a (sterilized) empty tips container to hold upright while working with
Total volume of leukapheresis product can vary considerably between donors/collection but typically you’ll receive 40-80 mL in a quarter leukopak
3. In biosafety cabinet, with full PPE & using sterilized scissors, cut off top of leukopak and discard this plastic in biowaste bag.
Even though donors are screened for particularly nasty viruses (HIV-1/2, Hepatitis B/C) within 90 days prior to donation, we will treat as if contaminated w/ infectious agent to be extra cautious
4. Add 100-200 uL of DNase I (5 U / uL from Zymo) directly to leukapheresis product and mix by pipetting up & down w/ P25 serological. Incubate contents 5-10 at room temp.
It’s likely some cells lysed in the ~24h since collection, during ambient transit, especially fragile neutrophils. Even if only 10% of all neutrophils popped, the amount of gDNA released could significantly increase the viscosity of the sample and disrupt downstream steps (3E9 nucleated cells * 4% neutrophils * 10% lysed = 12e6 cells * 6pg gDNA / cell = 72 ug of long stringy DNA released into solution
To combat, I recommend stocking DNase I from Zymo (or any other manufacturer) suspended at the recommended concentration of 5 U / uL.
Applying 100 uL per 40 mL of leukapheresis product = 500 U of DNase I which should be a saturating amount
According to Zymo, 1 U of their DNase I should be enough to degrade 15 ug of DNA in <10 min at 25C, HOWEVER, this is assuming a sufficient concentration of calcium ions (cofactor)
Since our leukapheresis product was mixed w/ ACDA to prevent coagulation, it’s likely calcium is very limited in solution, hence the copious amounts of DNase I added. I chose the total amount to be proportional to Zymo’s recommendation for degrading undesired gDNA in their RNA purification kits: 6 uL of 5 U/uL Dnase I (=30 U total) to degrade gDNA from 1E6 cells in 10-15 min at room temp. (on column for Direct-Zol kits, which I prefer). So 30 U / 1e6 cells * 12e6 cells = 360 U (rounded up to 500 U to be safe).
5. While incubating, prepare 50 mL conical tubes for density separation by aliquoting 15 mL of Lymphoprep (density gradient medium from STEMCELL) per tube (n=4-8 tubes total)
You can alternatively use Ficoll or Ficoll-Paque just note their specific density & centrifugation instructions
These blood cell separation mediums are all just a mixture of a highly branched polysaccharide (sugar polymer) and sodium diatrizoate in water, carefully measured to achieve a density of 1.077+/-0.001 g/mL
Actually, Ficoll is just the polysaccharide, however it’s less ideal to work with b/c it’s highly viscous and hard to aliquot. This syrupy property also prevents cells from separating cleanly when spun.
Diatrizoate is heavy small molecule w/ n=3 iodine atoms to increase density w/o raising the viscosity of the solution as much. Also, the diatrizoate helps maintain osmolarity at ~290 mOsm (“milli-osmolar”) to keep cells in their natural state (as opposed to swollen or shriveled or lysed). Note: 1 mOsm = 1 mM of dissolved solutes = 1E-3 moles of dissolved solutes per L of solvent
6. After 5-10 min of DNase I treatment, transfer 10 mL aliquots of treated leukapheresis product to separate 50 mL conical tubes (not the tubes containing Lymphoprep yet)
7. Add 21 mL of room temp. EasySep buffer per tube (i.e. dilute leukapheresis product 0.3x). Invert to mix. This step is to chelate any free calcium to fully stop the DNA digestion rxn.
8. Now, SLOWLY & CAREFULLY, aliquot 30 mL of diluted leukapheresis product (the full amount in each conical tube) to the 50 mL conical tubes containing Lymphoprep
This step is one of the trickiest & most finicky. You want to layer the cells gently on top of the separation medium, being careful to maintain the barrier (“porous frit”) between them. Otherwise, clean separation won’t occur during centrifugation. I highly recommend trickling the cells down the side of the tube to achieve this as opposed to adding dropwise. You can purchase specialized barriers to help simplify this annoying step of the protocol. I’ve never used them but I’m tempted every time I do this (as my shoulder starts to get sore from holding everything up to eye level to make sure I’m maintaining the barrier). All in all, takes 5-15 min just to layer cells into the tubes
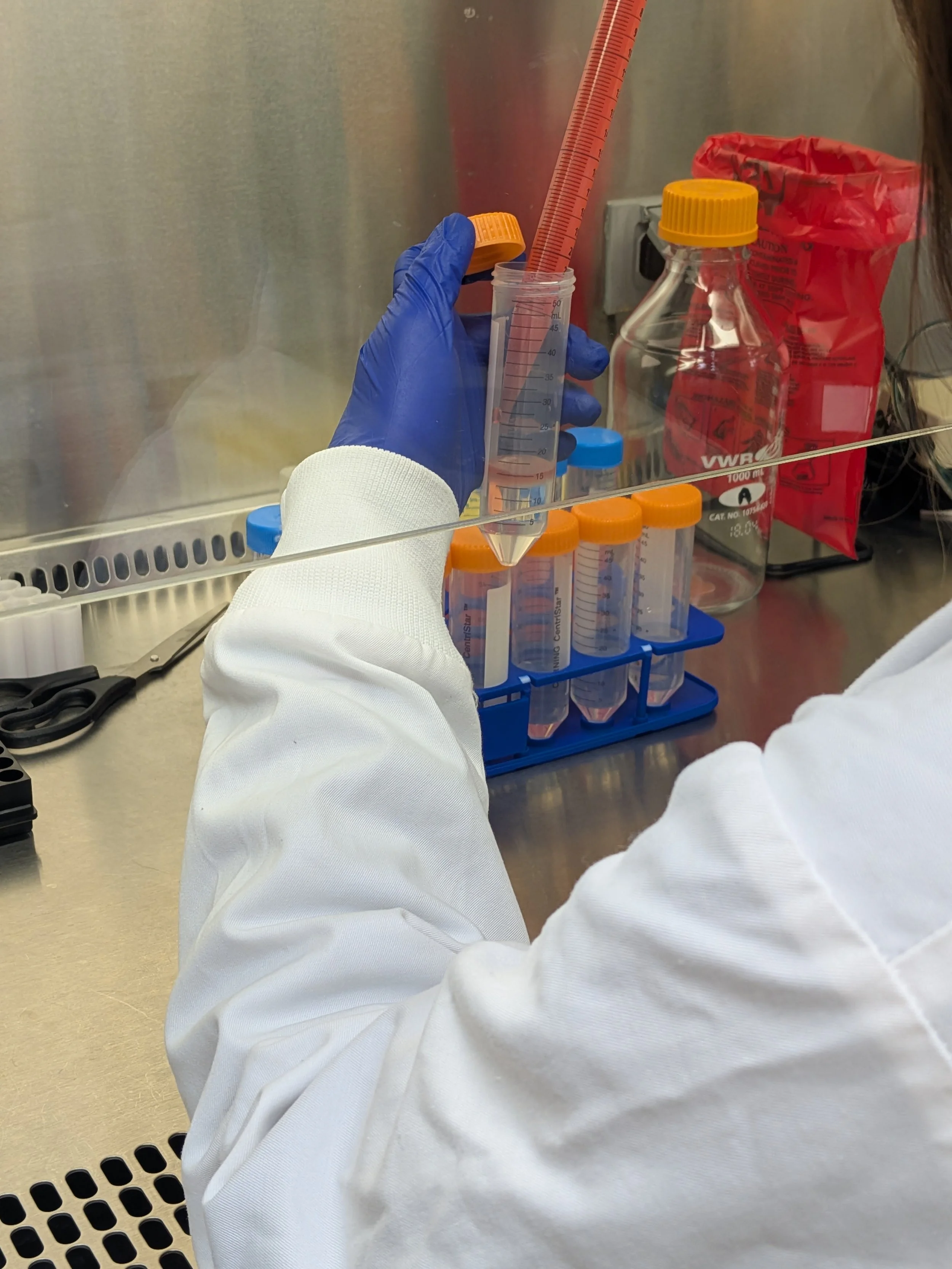
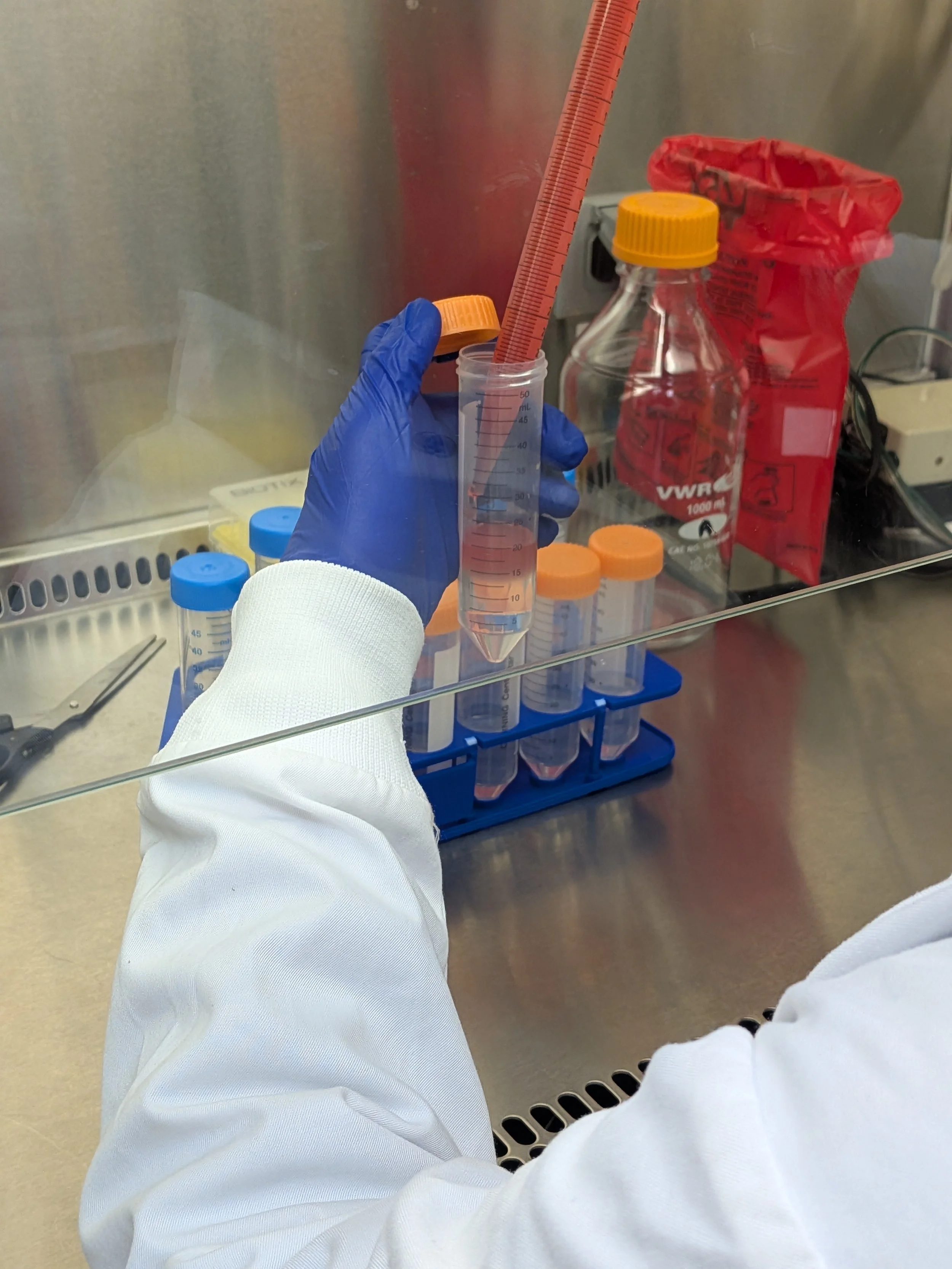
If you don’t want to do a tricky step, have an undergrad do it! You know, for training purposes…
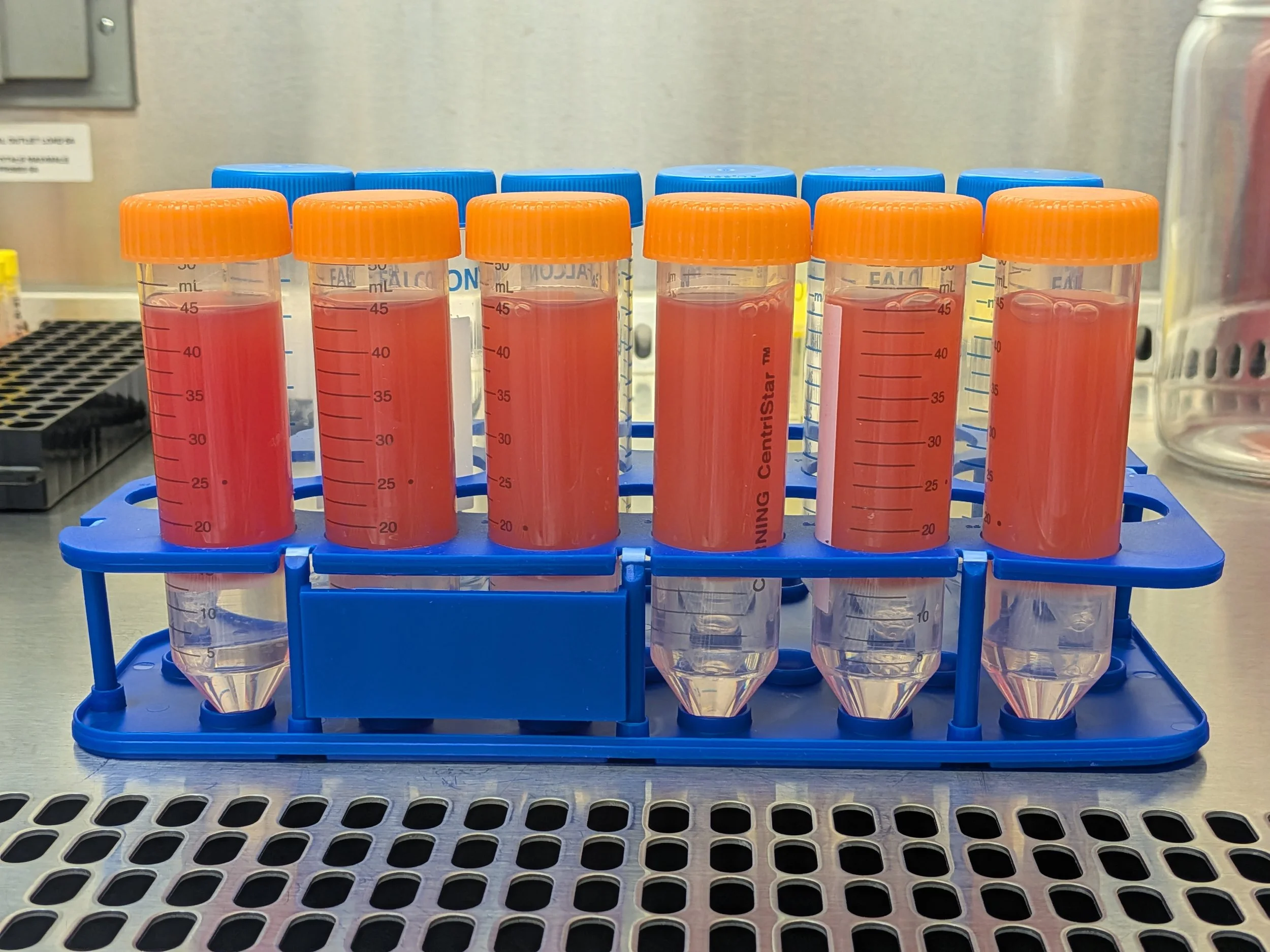
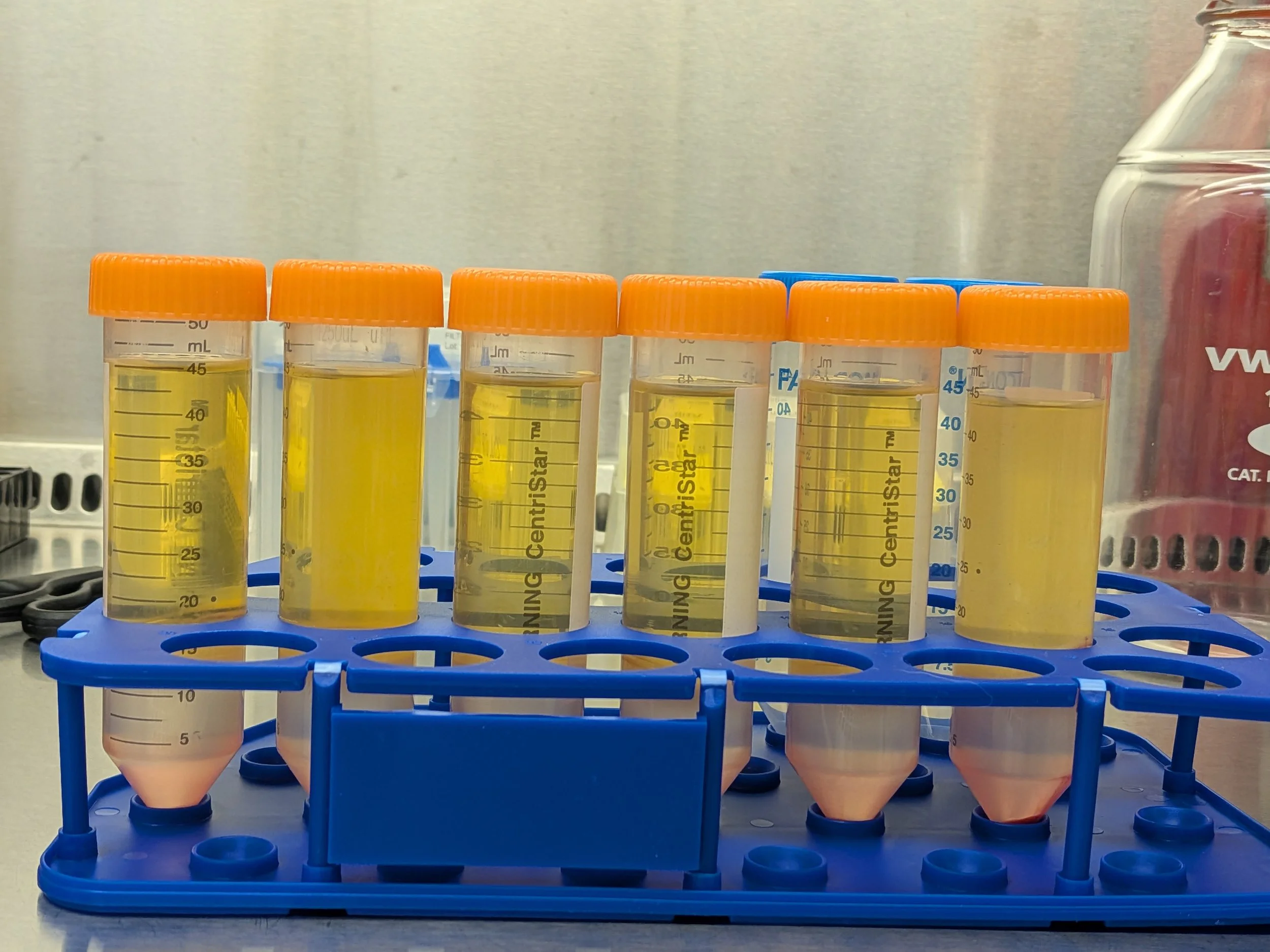
Above) Pre-spin, beautifully layered Below) Post spin
9. Once done layering the cell suspension on top of lymphoprep aliquots (now 45 mL / tube, n=4-8 tubes), spin 800xg for 30 minutes w/ brake off (use swinging bucket, takes ~36 min total)
10. Once done spinning, slowly & carefully lift tubes to confirm distinct, separated layers then transfer tubes back to biosafety cabinet taking care to maintain layer separation
From top to bottom: Plasma (transparent yellow liquid, ~30 mL total), PBMCs (thin, cloudy, bouncy white layer, ~5 mL total), lymphoprep (hazy clear layer, ~10 mL total), red cell pellet (RBCs & granulocytes, very bottom)
11. Carefully remove plasma layer without disturbing cloudy PBMC layer. Discard plasma into waste
Draw from top of liquid line. Don’t need to get every last drop of plasma layer here, can leave a little behind to be safe
12. Collect thin, cloudy “buffy coat” PBMC layer (and a little bit of cushioning lymphoprep)
Should recover 5-10 mL total per tube. Again, it’s okay if you get some of the adjacent layers b/c we’ll wash the cells
13. Pool collected PBMC fractions together into single 50 mL conical tube
14. Dilute pooled PBMCs up to 50 mL total volume w/ room temp. EasySep buffer
Discard remains of gradient separation step (I usually pool into glass jar to bleach & discard at the end)
15. Split diluted PBMCs 2x25 mL into fresh 50 mL conical tubes labeled “T fraction” and “M fraction”
These separate batches (technical replicates) will be the inputs for the EasySep isolation kits
16. Dilute 10 uL aliquot from each replicate tube into 990 uL dPBS preloaded into Falcon flow tubes
17. Measure PBMC concentration by flow cytometer (we have an Attune Nxt in lab) or Countess
Flow 100 uL of 0.01x diluted samples @ 100 uL / min
See below for details on expected results


Removing top, yellow plasma layer (30 mL total)

Collecting thin, cloudy layer of PBMCs (“buffy coat”, 5-10 mL)
Pooling PBMCs from separate tubes together
What should we expect by flow?
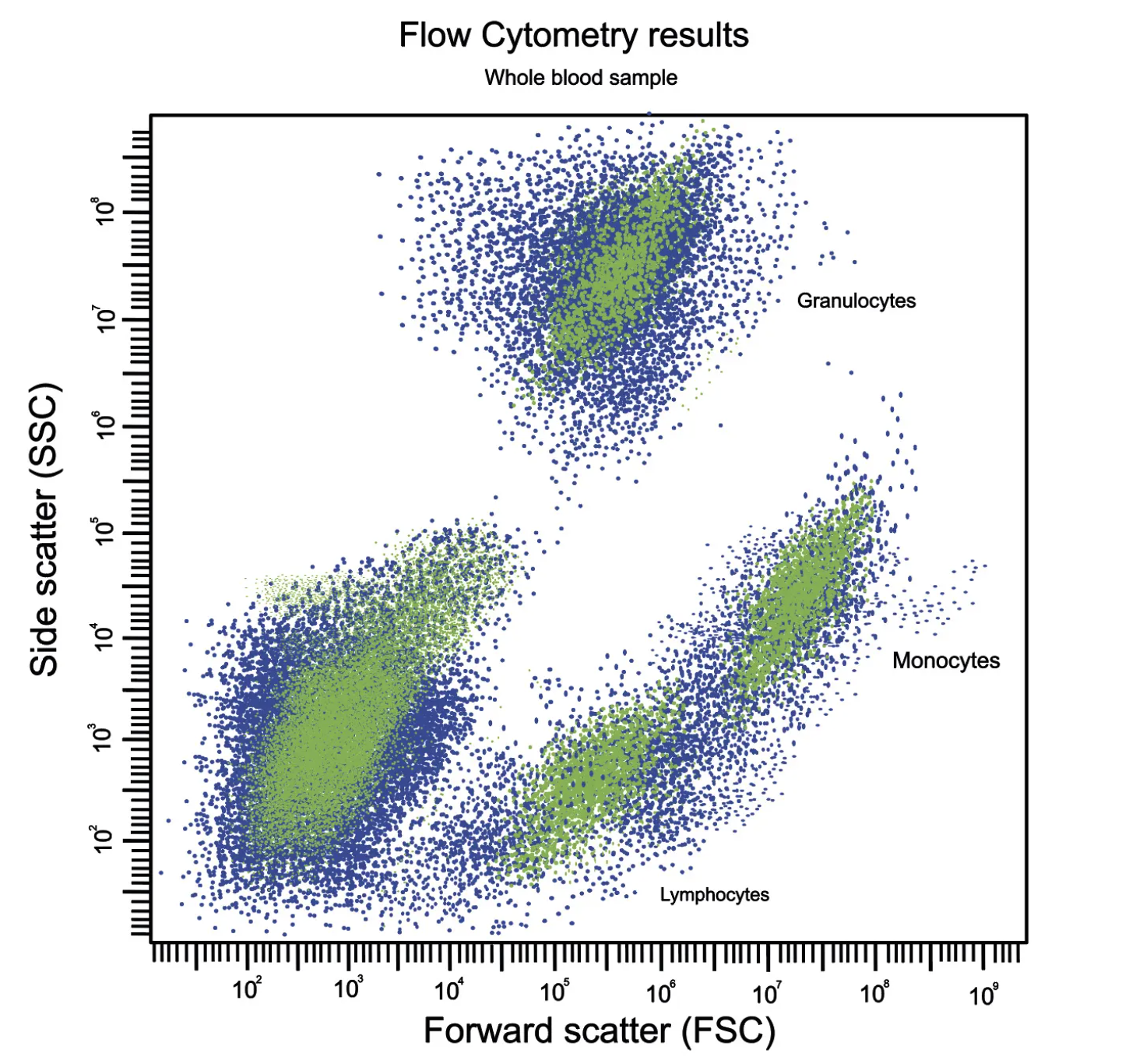
I think this figure on the left is an idealized/theoretical density plot of forward vs. side scatter but I like it b/c it gets the gist. Note the log scaled axes. The large population of events in bottom left is comprised of cell debris, platelets, and RBCs.
Lymphocytes are clearly distinguishable from background but are classically small, round cells hence the low FSC/SSC vals
Monocytes are the largest white bloods cells and slightly more irregularly shaped hence the higher FSC and SSC
Granulocytes are, by definition, highly granular (complex cytoplasm from all the protein packed granules & lobed nuclei) hence the very large SSC values but are roughly the same size as lymphocytes hence their overlapping position on the x-axis
Depending on how well preserved / intact your sample is upon arrival, these cell populations will be shifted around slightly compared to their idealized positions. The biggest difference you may observe is that populations shift left by FSC (cells get smaller) as they become stressed and start to shrivel up. This unhealthy/shriveling morphology also causes SSC to trend up slightly before the cells finally collapse into the debris field as they lyse / apoptose.
Note that these “dying trends” occur independently across the different cell populations (neutrophils are less robust than monocytes which are less robust than lymphocytes) usually starting around 6-12h post collection and continuing thereout
There’s a great paper tracking PBMC health over time by flow cytometry from 2022 (their main figure is shown below)
Their title & data speak for itself, basically neutrophils start dying and contaminating PBMC preps 6-24hrs post collection while monocytes start dying after 24-48 hrs. So long as you can get them before that though, cells appear to somewhat maintain their morphology & primary functions.
https://clinlabint.com/the-vast-uses-and-advancements-in-flow-cytometry/
Johnson, R.K., Overlee, B.L., Sagen, J.A. et al. Peripheral blood mononuclear cell phenotype and function are maintained after overnight shipping of whole blood. Sci Rep 12, 19920 (2022).
Note: blood was stored/shipped at ambient temp. post collection
How about our data?
Keep in mind that, yes, it would be great if we could immunostain and clearly distinguish/quantify cell populations by their surface markers but there’s no time when isolating!
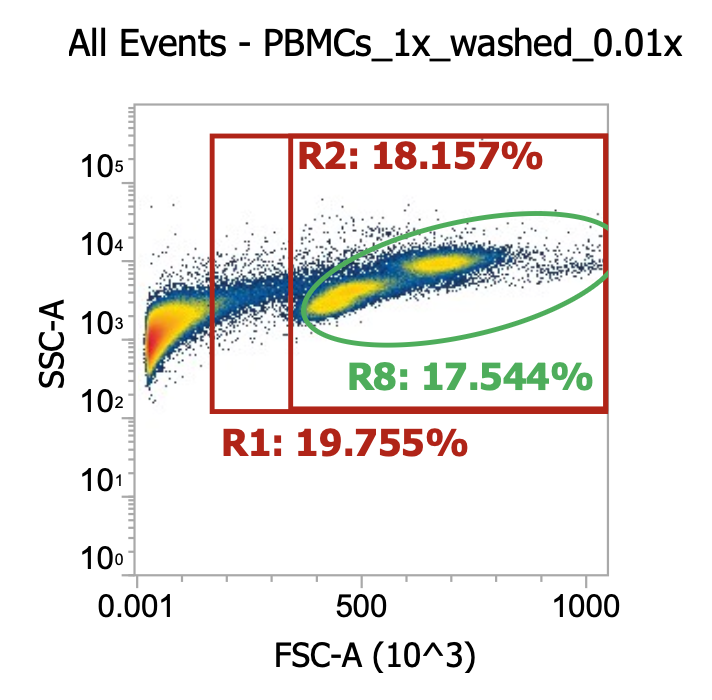
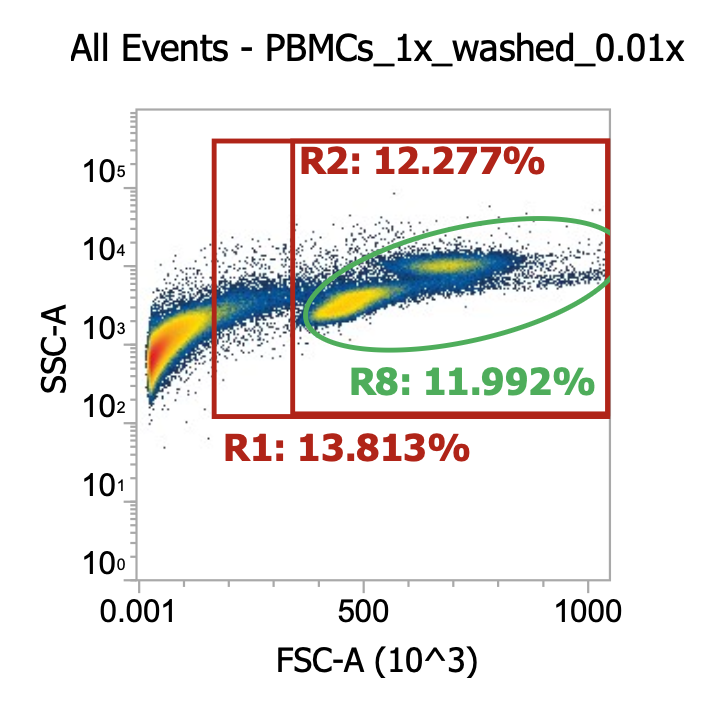
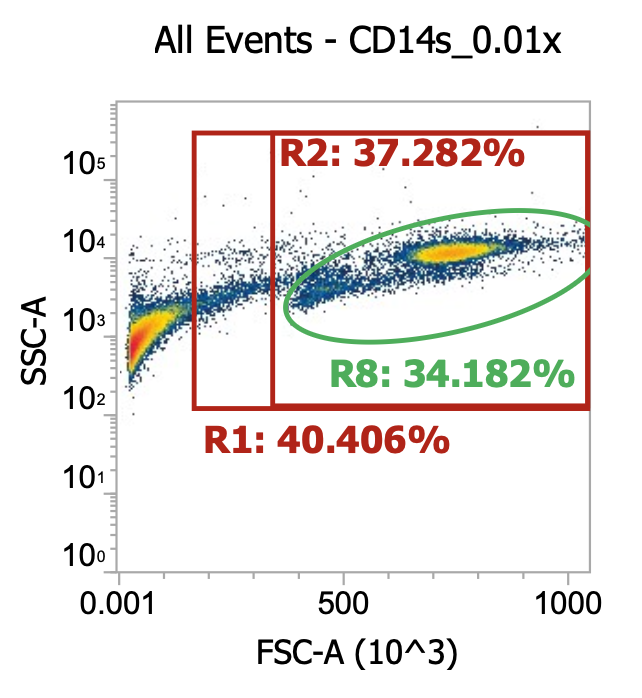
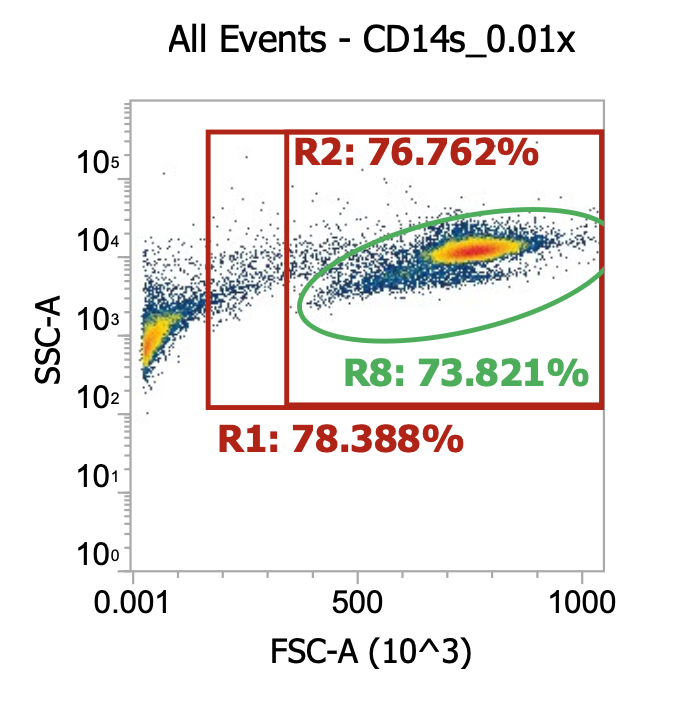
Two examples of “raw” leukapheresis product from different donors, diluted 200x
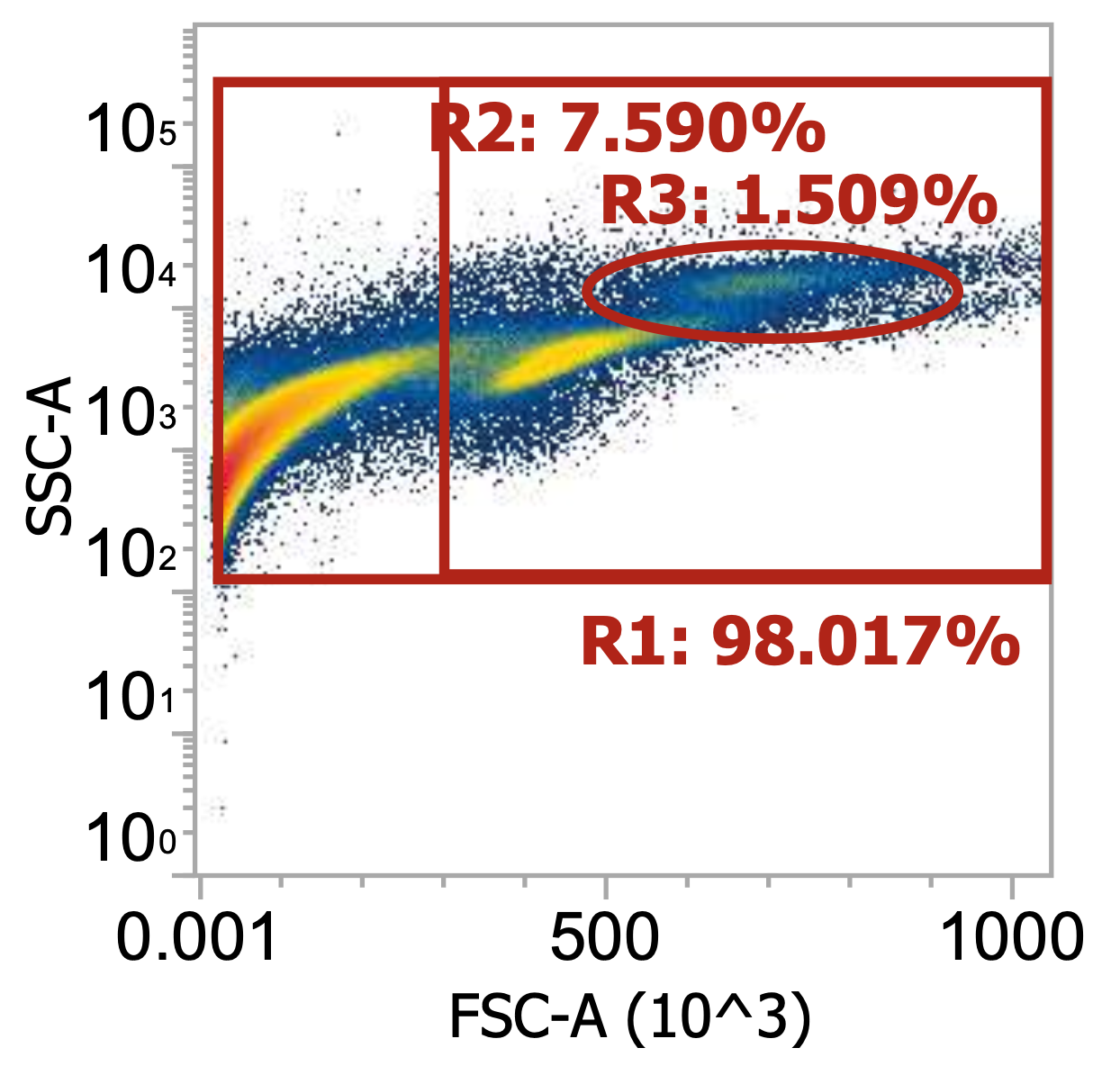
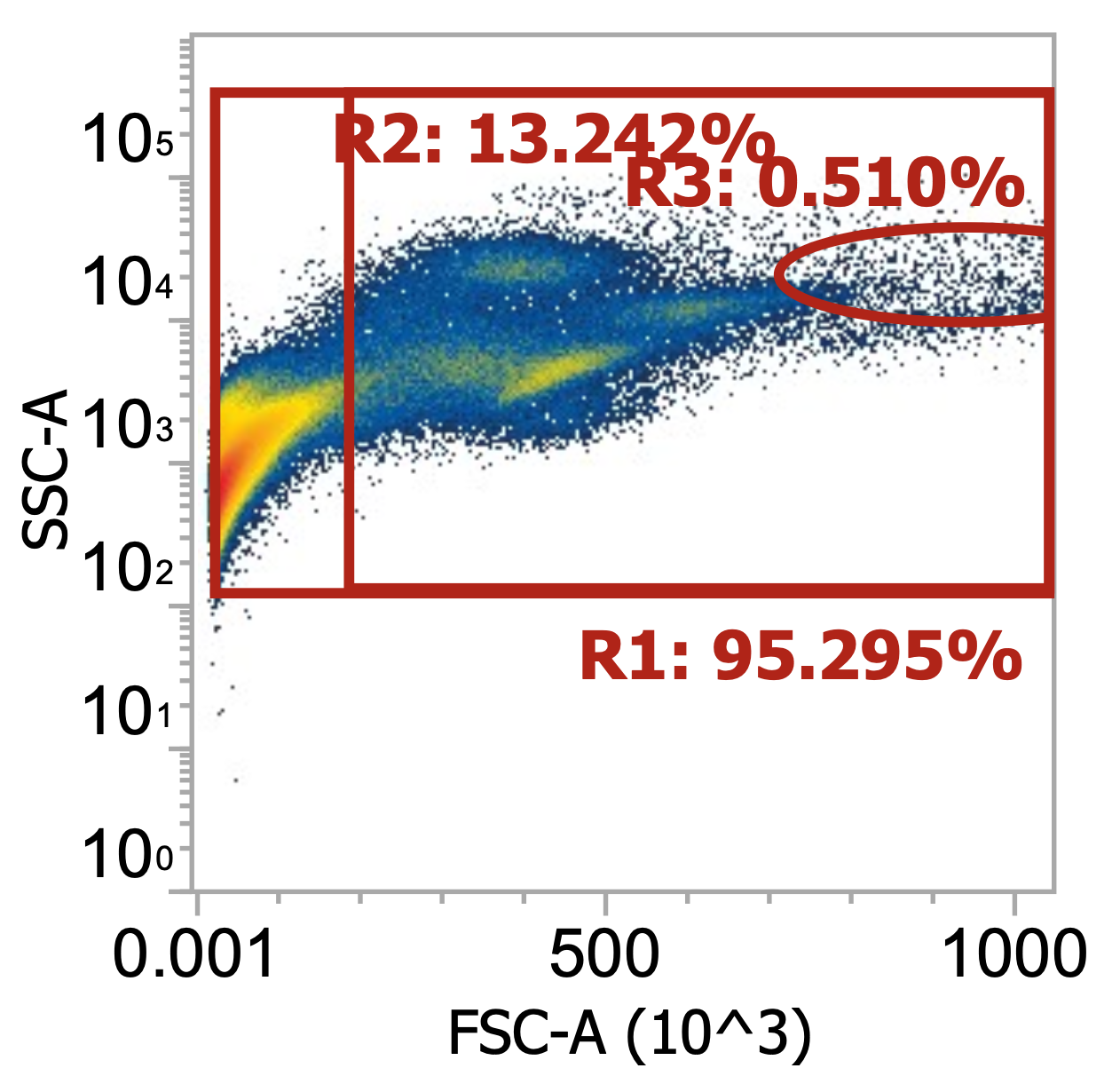
Ignoring my gates, you can clearly see the “debris field” (leftmost population) and somewhat make out the lymphocyte and monocyte populations in each donor. For the donor on the right, you can also see a distinct population of granulocytes (top cluster). Hopefully, you get a sense of how “dirty” and variable leukapharesis product can be.
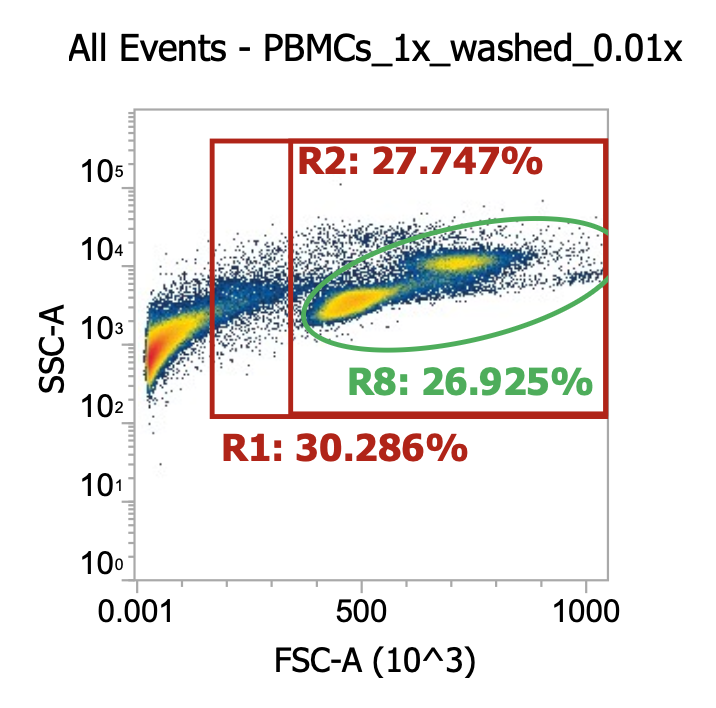
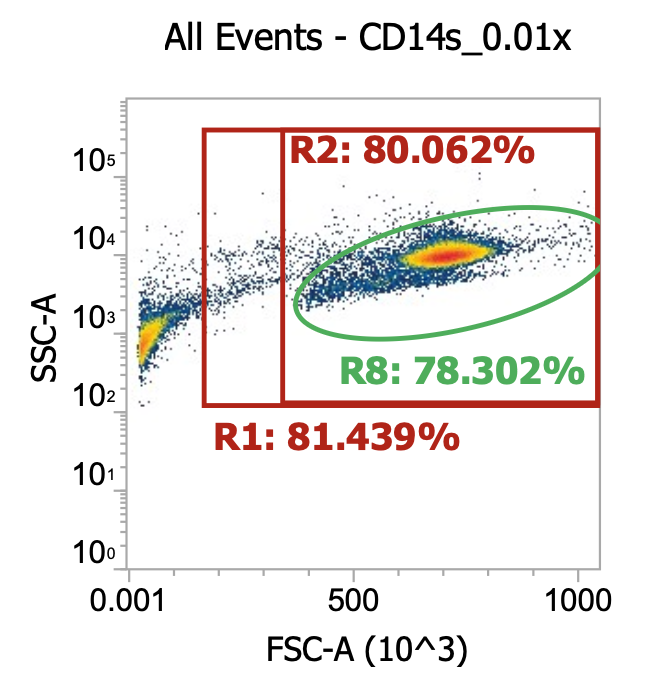
Three examples of PBMCs isolated from different leukopaks, diluting collected buffy coat 100x
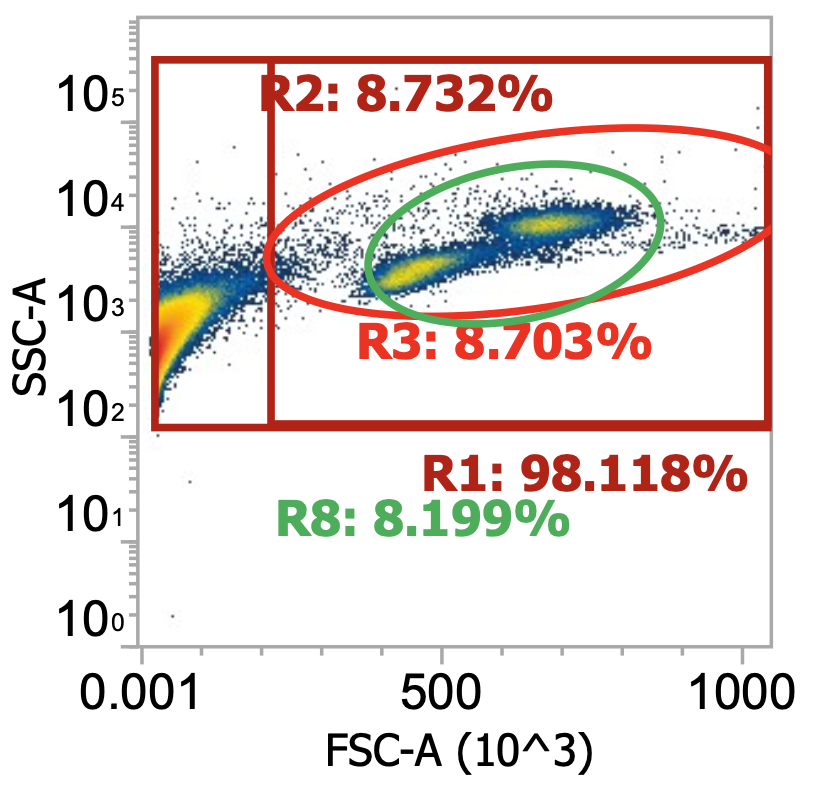
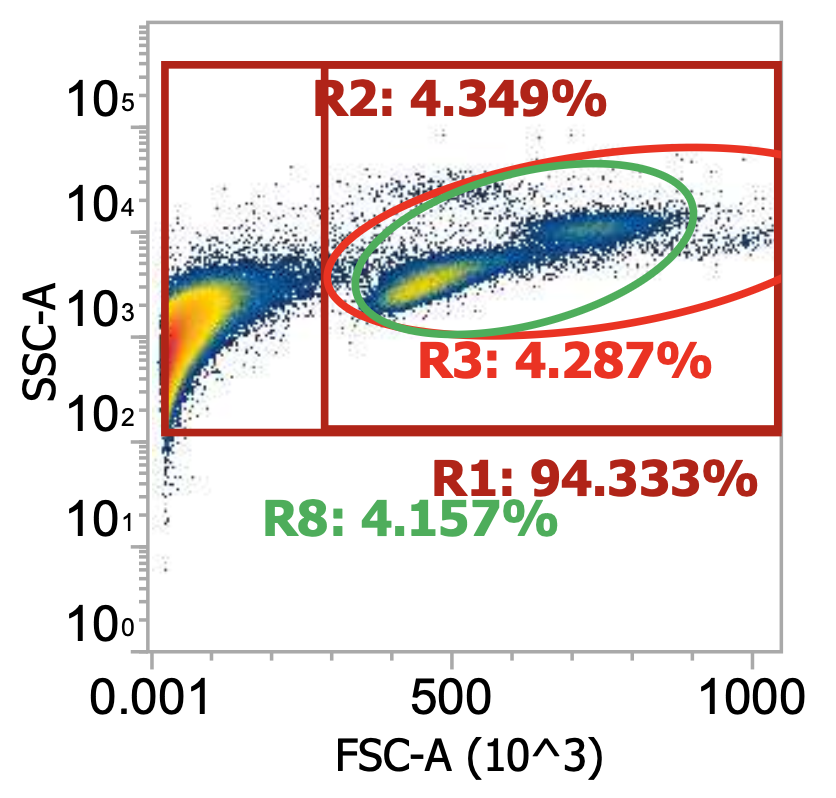
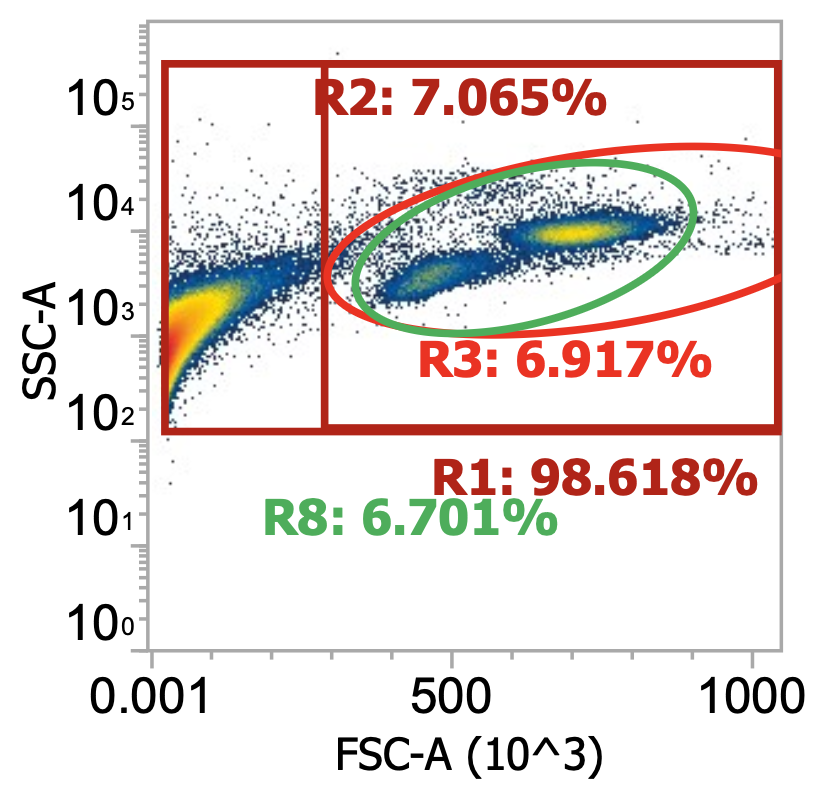
Above, the smaller lymphocyte and larger monocyte populations (by FSC) are much more distinguishable from each other and the background debris. However, even in these “pure” samples where PBMCs have been enriched by density gradient, target cells only represent 4-9% of the total events detected (much junk remains esp. platelets).
Note the variability in the lymphocyte-to-monocyte ratio across donors by relative cluster densities (in order, roughly 1:1 vs. 2:1 vs. 1:2). Blood composition is dynamic.
Great, we’ve verified our target cell types are enriched in the density separated PBMC-fraction of leukapharesis product. Now what? As noted above, our samples are still quite “dirty” with mononuclear cells constituting <10% of total events detected by flow (i.e. the samples are dominated by cell debris and platelets). Therefore…
Part 3: Wash PBMCs prior to isolating target cells w/ EasySep kits
I’m going to focus on a specific experiment (processing “Donor 9” leukapheresis product) from here on out so that I can provide hard #s while ensuring the data remain consistent & directly comparable across steps.
1. Spin both tubes of collected PBMCs 200-300xg for 10 min w/ brake on (I recommend 250xg with a swinging bucket rotor)
2. Confirm large, white cell pellets. Then, transfer 45 mL of supernatant per tube to labeled 50 mL conical tubes (“wash 1 sup.”) then resuspend cell pellets to 50 mL total w/ EasySep buffer.
I highly recommend adding ~20 mL of buffer then flicking bottom of tubes to get cells back into suspension before adding the remaining ~30 mL of buffer. And/or pipette buffer gently up & down to resuspend pellet
We are saving the supernatant for now as due diligence to confirm later by flow that most cells were pelleted in the first spin (at 250xg they very well should be, I’ve spent lots of time optimizing this # to ensure both monocytes & T cells are pelleted gently w/ minimal losses). Other protocols may recommend 120-150xg to enrich for cells while depleting platelets but I’ve found these lower speeds always fail to pellet many of the desired WBCs
3. Spin cell suspensions again, 250xg for 10 min w/ brake on. Again, transfer sup. to separate, labeled conical tubes (“wash 2 sup.”) then re-suspend cell pellets in 20 mL EasySep buffer
This 20mL final volume per replicate ("T” fraction and “M” fraction) is optimal for input into the EasySep kits and should lead to a cell concentration near the kits’ recommended input concentration of 5E7 cells/mL (1E9 TNCs)
4. Dilute small aliquot of washed cells 0.01x in dPBS (10 uL of cells in 990 uL dPBS) and measure by flow cytometry (or Countess)
Below, I flowed 100 uL of diluted replicate samples (“T” fraction and “M” fraction) @ 100 uL / min. You can also dilute & flow the collected supernatant to be rigorous although usually you’re crunched for time
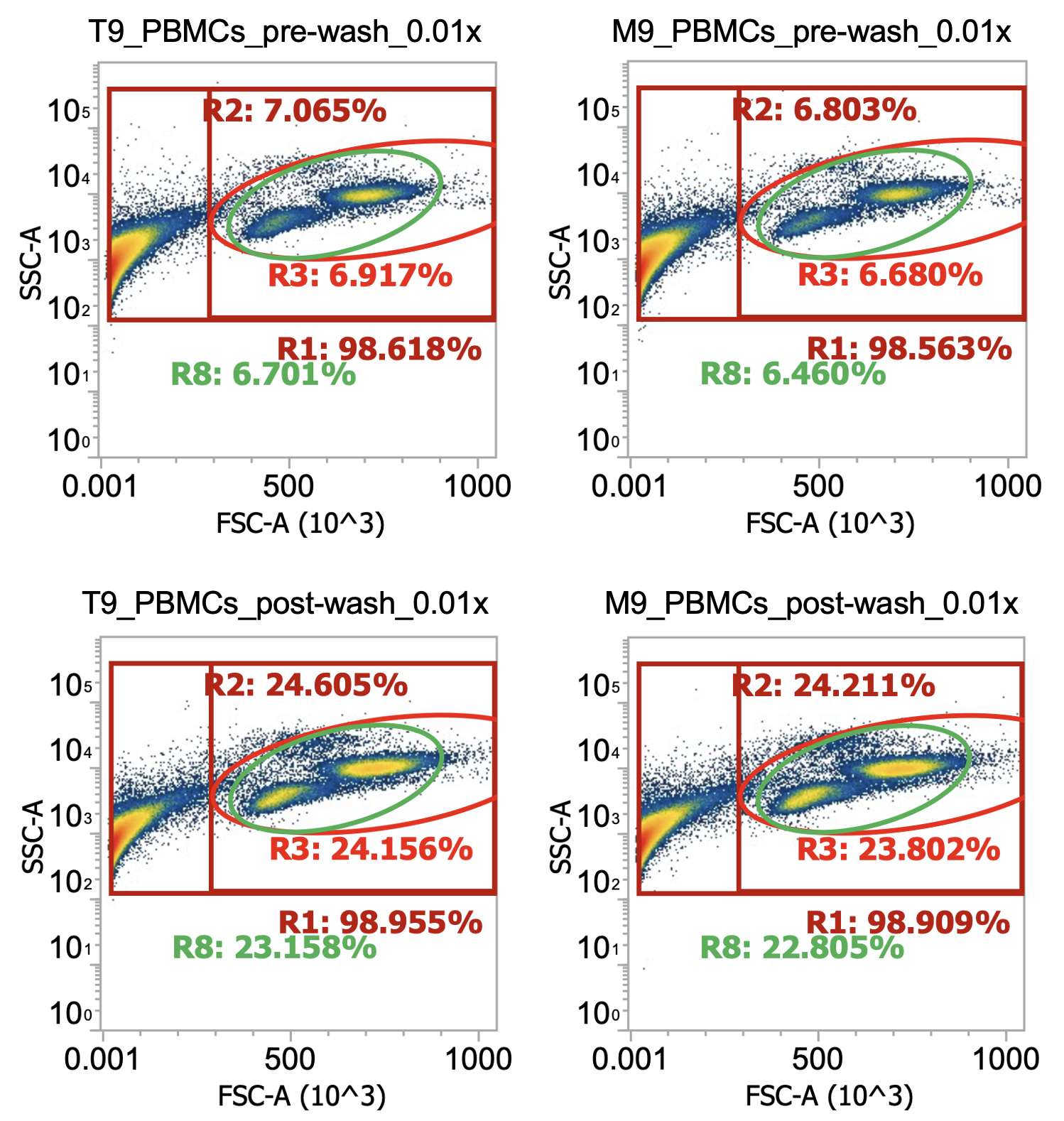
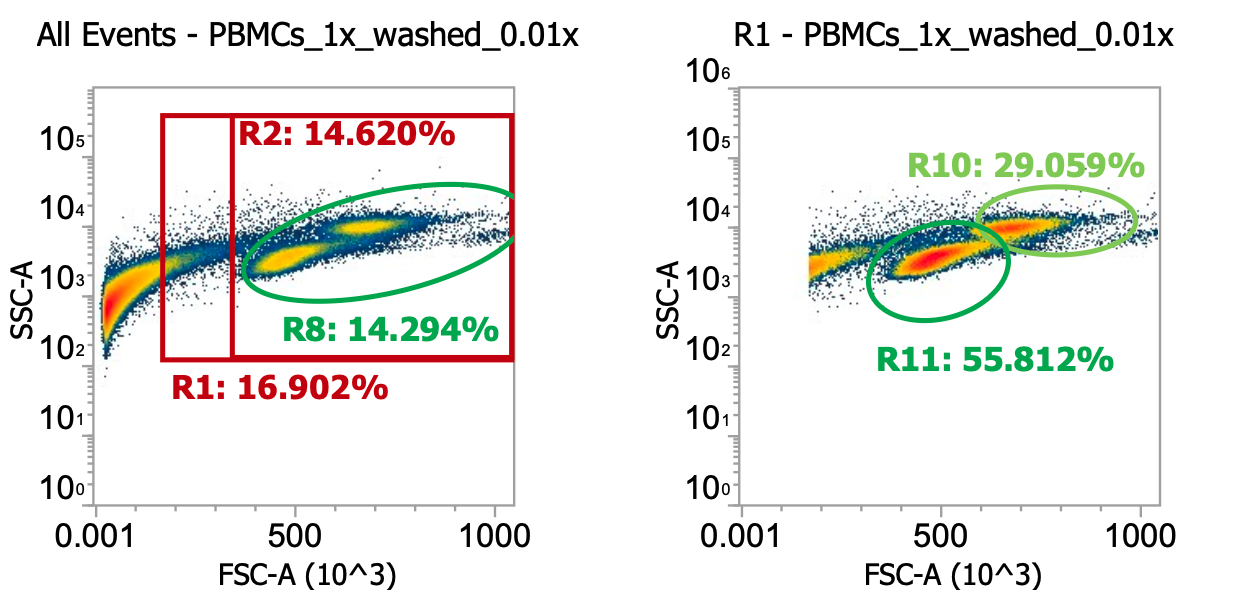
Comparing top and bottom rows of technical replicates, it looks like we’ve enriched our target cells 3.5x
(gated cells increase from 7% of total detected events to 24%) by washing twice at 250xg
What about total cell #s pre and post-wash?
PBMCs, T fraction, pre-wash) 1.7e7 cells / mL * 50 mL = 0.85e9 total nucleated cells (“TNCs”)
PBMCs, M fraction, pre-wash) 1.7e7 cells/mL * 50 mL = 0.85e9 total nucleated cells (“TNCs”)
So a total of 0.85e9*2 = 1.7e9 PBMCs recovered from density separation from a total starting amount of ~2.5e9 TNCs (according to supplier). I’ll typically recover 70-80% of total cells from the density separation
PBMCs, T fraction, post-wash) 3.7e7 cells / mL * 20 mL = 0.74e9 TNCs….. vs. 1e9 TNC target
PBMCs, M fraction, post-wash) 3.9e7 cells/mL * 20 mL = 0.78e9 TNCs…. vs. 1e9 TNC target
So a total of ~1.5e9 PBMCs recovered after washing (~90% recovery) confirms our centrifuge speed & time were sufficient to pellet the cells, although it also retained many of the platelets we were trying to remove
There is always this tradeoff in leukopak processing; spin slower and you increase cell purity but decrease total cell recovery (and vice versa). The real question is, how low of purity (i.e. how many platelets) can the EasySep kits tolerate? The answer is: these samples (roughly 25% PBMCs, 75% platelets/debris, at a final cell concentration of ~4E7 cells/mL) are sufficient.
Note: You can actually see the monocytes starting to grow unhappy in the bottom row of plots (post-wash) as a small/loose cluster of cells shifts slightly up & to the left of the primary monocyte population.
This is a good reminder that time is of the essence and we should finish isolating / freezing down these cells ASAP
What if we spun at slower speeds?
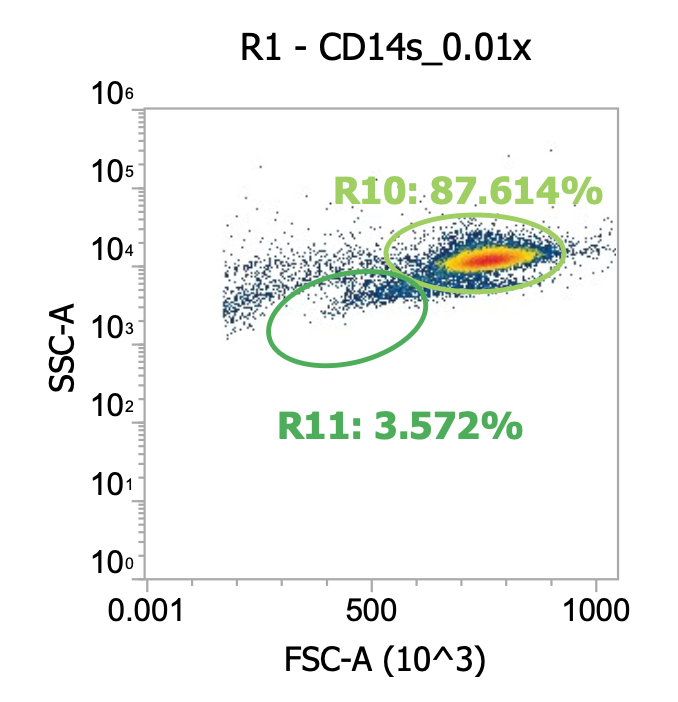
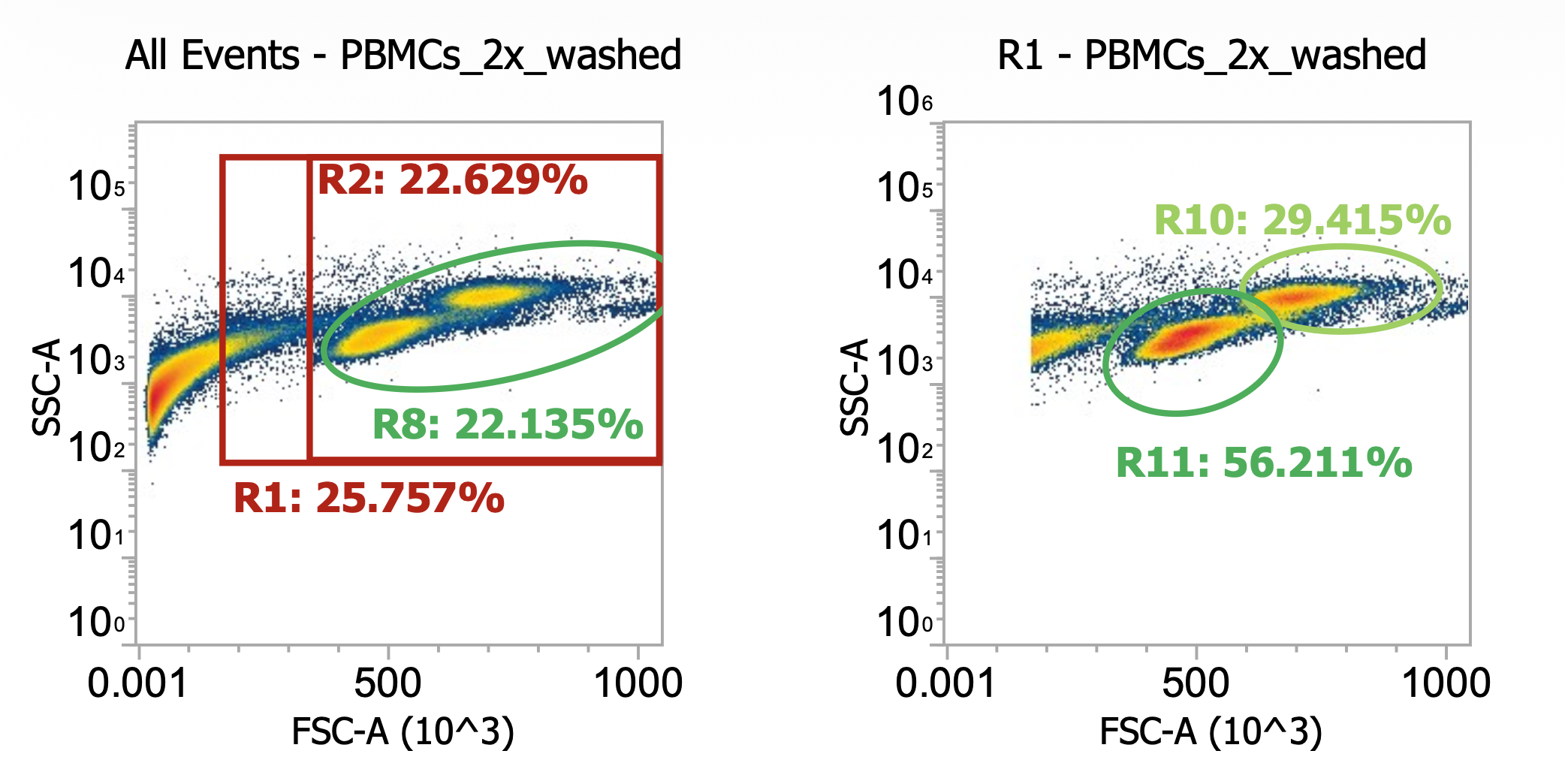
Fortunately, I have that data! When processing “Donor 9”, I actually initially washed the collected PBMCs at 120-150xg (don’t remember which, oops) for 10 min w/ brake off. Afterwards, I noticed my cell pellets were kind of small and the collected supernatant (“sup.”) was pretty white/cloudy suggesting that a lot of the cells failed to pellet so I diluted and measured the sup. by flow before spinning both the sup. and resuspended pellets 250xg for 10 min w/ brake on for “Wash 2” (I then re-combined the pelleted cells from both tubes). Here’s the comparison:
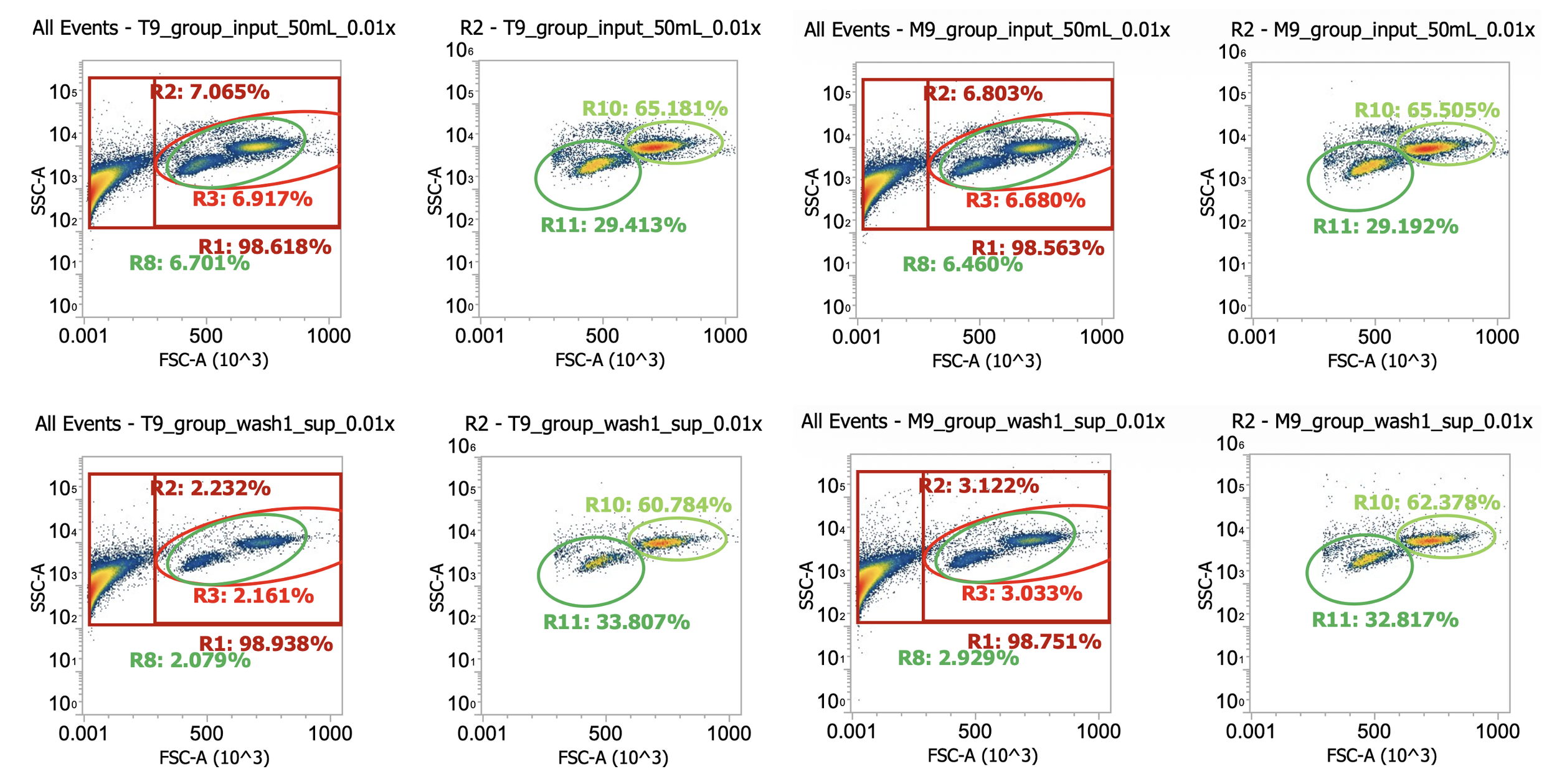
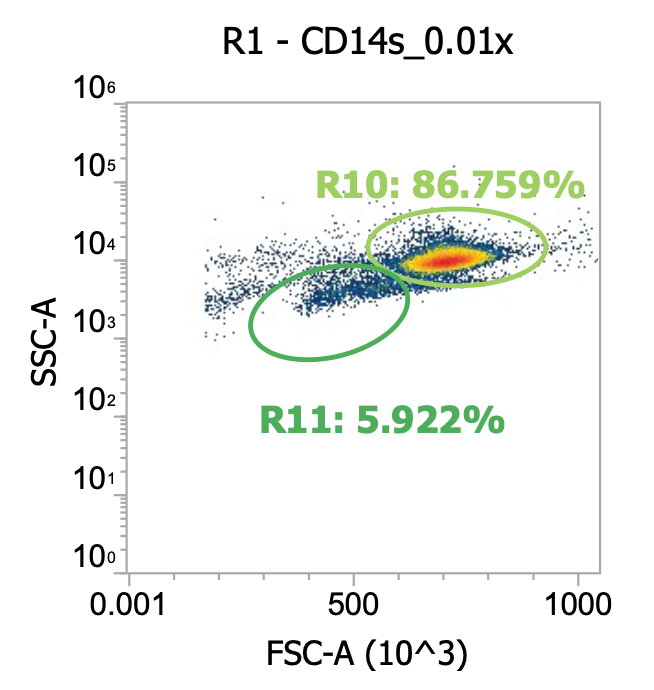
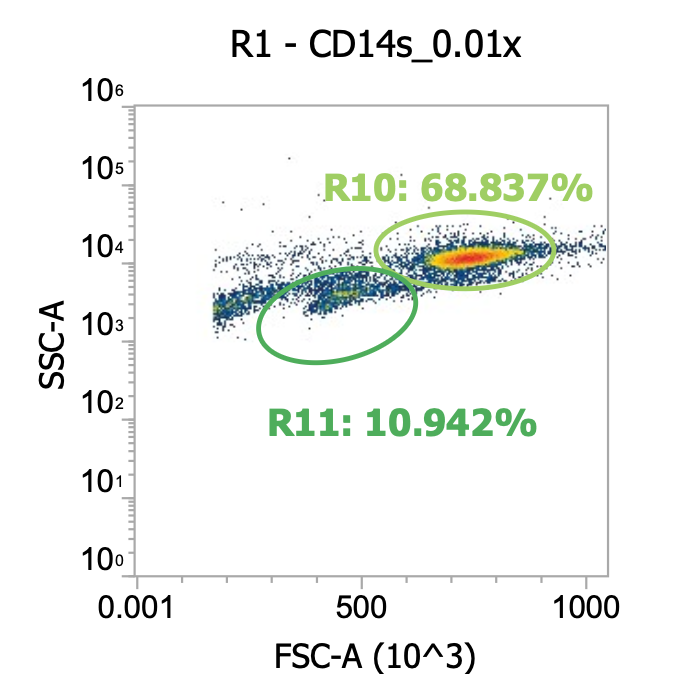
Okay here I am showing you the input into the wash step (top row, PBMCs) and the collected supernatant (bottom row, all cells not pelleted) after wash #1, diluted 100x
I’m including two plots per sample, the left plot shows all events while the right plot shows only the events (cells) in rectangular gate “R2”
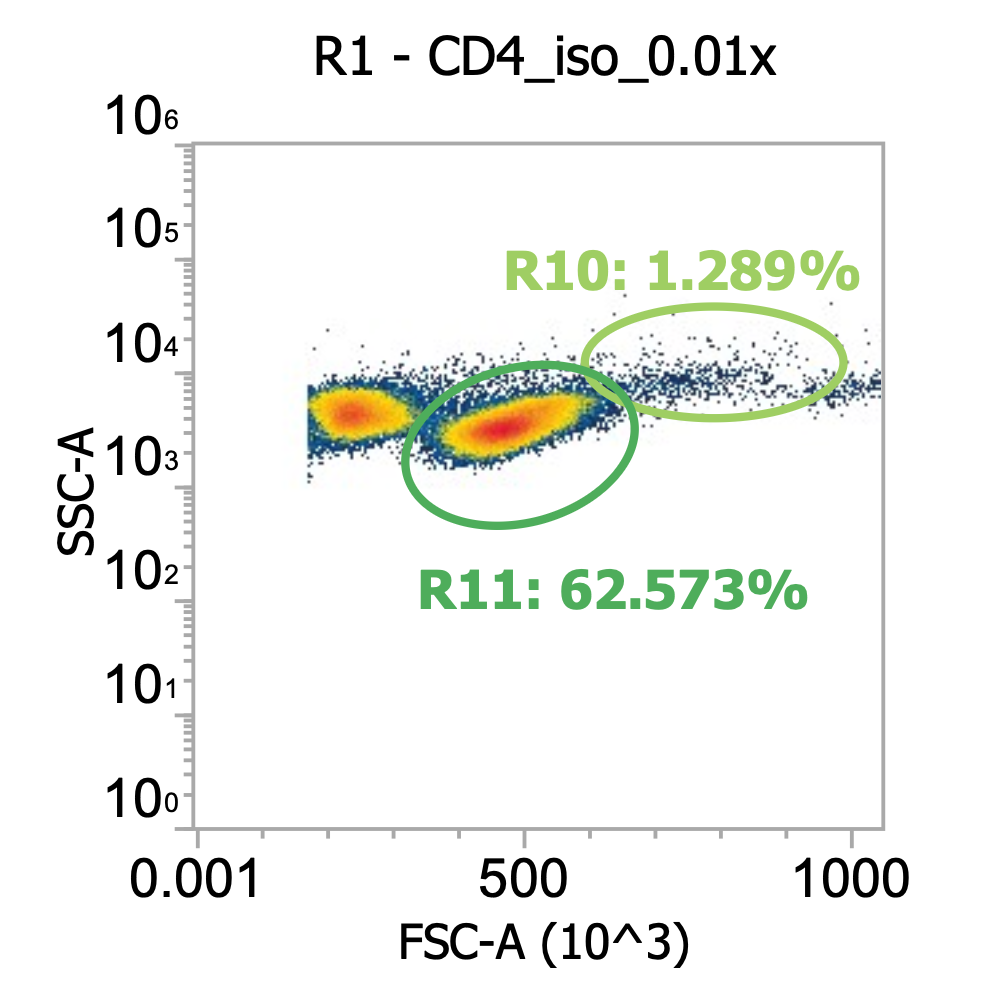
I’ve included additional oval (green) gates in the plot of R2 events to quantify the relative percentage of monocytes (R10 gate) to lymphocytes (R11 gate)
You can see, initially, we have approx. 65% monocytes and 29% lymphocytes. After spinning slowly (120-150xg), we have 61-62% monocytes and 33-34% lymphocytes.
This means the larger monocytes pelleted slightly better than the smaller lymphocytes (slightly counterintuitive) but only negligibly so - a significant amount of both cell types remain in the sup. (failed to pellet) after spinning slowly for 10 min
Results of spinning PBMCs <=150xg for 10 min w/o brake? Roughly 1/3 lost!!!*
T9_group_wash1_sup_0.01x) 4.5e4 cells/mL * 100x dilution * 45 mL ~= 2.0e8 TNCs … vs 0.85e9 input (i.e. ~25% of cells remain in sup/failed to pellet)
M9_group_wash1_sup_0.01x) 7.4e4 cells/mL * 100x dilution * 45 mL ~= 3.3e8 TNCs … vs 0.85e9 input (i.e.~40% of cells remain in sup/failed to pellet)
*Note the variability between technical replicates.The takeaway? Washing PBMCs at slow speeds can help clean up your samples (increase cell purity prior to immunomagnetic selection), however, take care in retaining your cells
200-300xg is the sweet spot, I recommend 250xg (with swinging bucket!)
Step 4: Isolating target cells by immunomagnetic selection (using EasySep kits from STEMCELL Technologies)
We’re finally ready to purify our desired immune cells (here, CD3+CD8+ T cells and CD14+CD16- monocytes, separately). In the past, I’ve tried running the two EasySep isolation kits side-by-side (in parallel) to minimize the amount of time it takes, however, I do NOT recommend this now. Turns out, following the exact incubation times recommended by STEMCELL’s protocols is critical to obtain the expected/advertised yields. Why Ryan? The kits are antibody based so worse case scenario isn’t incubating for longer better? It’s tempting to think so but, in fact, this couldn’t be further from the truth. Here’s how it works:
STEMCELL’s EasySep kits use “tetrameric antibody complexes (TACs)” to coat cells with small paramagnetic particles
An observed trend in molecular biology: if a synthetic spherical thing is micron scaled, or larger, it’s a ‘"bead”. If nano-scale then “particle”
TACs are two antibodies (usually different mouse IgG1s) fused together using two other partial antibodies (rat anti-mouse IgG1s F(ab')2 fragments). The bridging rat antibody fragments bind the Fc domains of the two mouse IgG1 antibodies to connect them and are likely produced by pepsin treating the full rat antibodies (to cleave off their own Fc domains).
Overall, it’s a very clever design that produces a bi-specific “molecule” that’s, otherwise, inert. Each end specifically binds to a different target antigen while the often-reactive Fc regions are either bound & blocked or removed entirely.
Many blood cells have general Fc receptors that bind the constant Fc ends of antibodies & induce a cellular response. Also, certain complement proteins in blood (part of the innate immune system) are design to react w/ Fc domains and trigger a cascade of chemical events that often lead to cell lysis.
Essentially, TACs = antibody connected antibodies
In the kits, one end (antibody) of each TAC binds to a cell surface protein specifically expressed by one of the blood cells we do NOT want to keep (hence the negative selection) while the other end (antibody) binds to the paramagnetic particles which are uniformly coated with a different antigen (e.g. dextran, a polysaccharide).
So really each EasySep kit contains a mixture of different TACs designed to target cell surface proteins unique to the cell types we want to remove
I doubt STEMCELL advertises their target proteins but you can assume they’re the usual markers (e.g. CD19 for B cells)
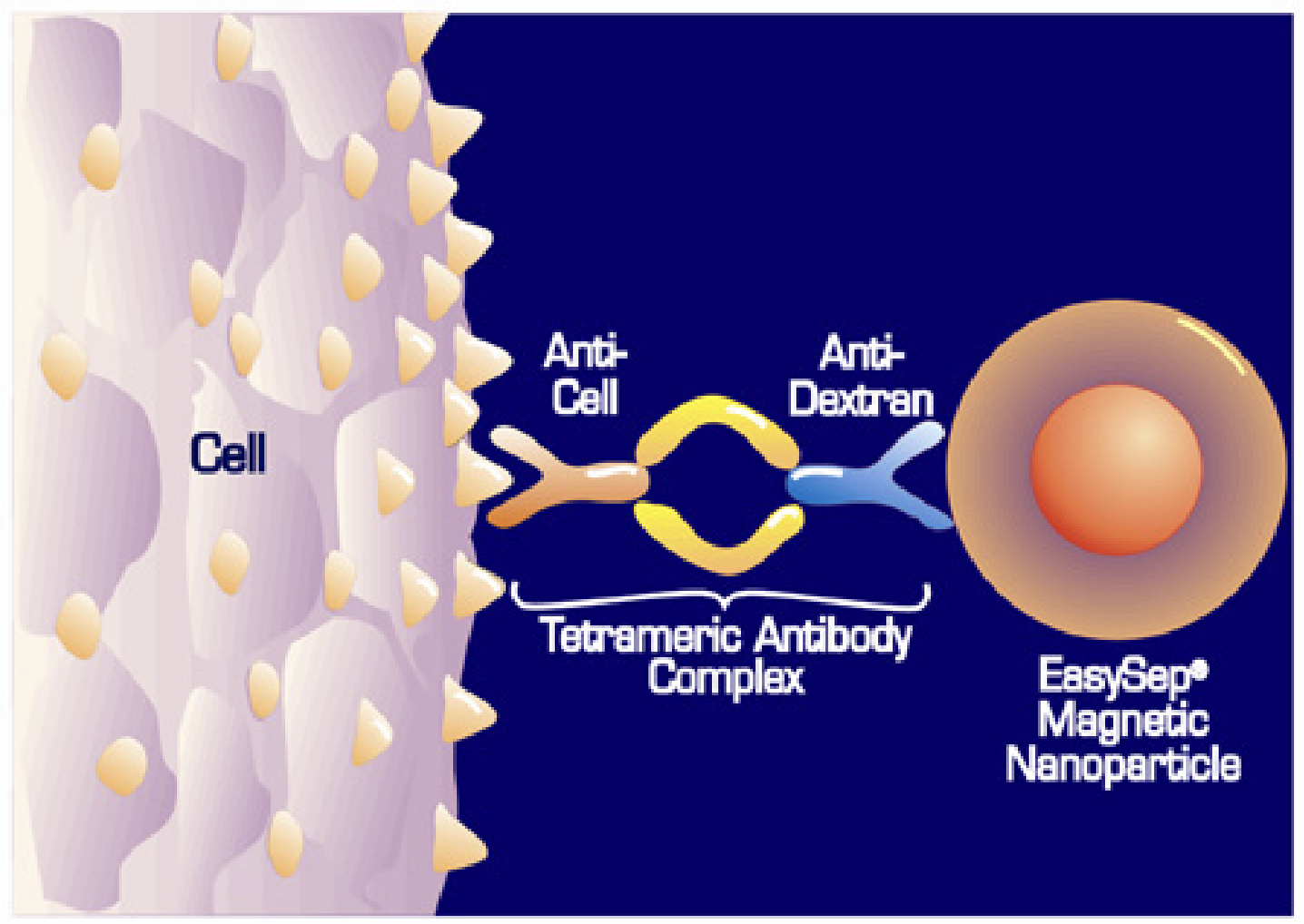
Sorry, this is the best pre-existing TAC binding cartoon I could get from STEMCELL. There are other diagrams but they highlight a different, indirect binding mechanism that combine TACs with another, labeled primary Ab.
Note: the paramagnetic particles are ~50 nm in diameter (~200x smaller than cells) and coated in antigen
So now hopefully the protocol becomes clear. For each kit we:
(1) add the TACs to our processed PBMCs, mix, and incubate, then
(2) add the paramagnetic beads, mix, and incubate, then
(3) apply the solution to a strong magnetic field, incubate, then
(4) collect the supernatant (unbound fraction of cells) to recover our final, purified population of negatively selected cells
The advantages of negative selection is that our cells of interest never bind any foreign TACs or particles minimizing the chances that they’ve become activated or responded in any way that affects their cellular state/behavior
Now, why do incubation times (and volume) matter so much? For this, it helps to imagine ourselves inside the tubes as the isolation takes place…
Our solution of cells, TACs, and particles is incredibly concentrated. As the TACs bind their target antigens, coating cells w/ paramagnetic beads, small lattices (three dimensional chains) of cells + particles quickly start to form since many particles can bind the same cell and vice versa. These cross-linked structures of cells+particles quickly grow larger and larger in solution and start to nonspecifically trap other cells including our desired (non-coated) cell type. This is especially pertinent when the magnetic field is applied as these large, floating meshes of TAC-bound cells+particles (& the unbound cells trapped within them) are dragged through solution towards the inner walls of the tube, co-precipitating (trapping) more free floating cells against the tube along with them. Sound too imaginative? These are documented effects known as “TAC crosslinking” and “cell entrapment” and they are incredibly time dependent! Incubate cells+TACs+particles for just the right amount of time (3-5 min) and the TACs bind their intended targets w/o forming many larger, cross-linked clumps thus minimizing any free floating cells from being co-precipitated once the magnet is applied thus recovering the expected yield of target cells. Wait just a little longer (5-10 min) before applying the magnet and your cell recovery drops from 70+% of expectation to <10% suggesting the cross-linking effect increases non-linearly (accelerates w/ time) - this matches the physical principles described in polymer chemistry (“branched networks”) and flocculation kinetics known generally as the “snowball effect” w/ an added layer of complexity introduced by the magnetic field
This would be a cool system to simulate with agent-based modeling to further characterize the kinetics (and critical parameters)
While I’m pretty confident this snowball dragnet effect is the main phenomenon negatively affecting yield in the EasySep kits, some other things may be exacerbating the effects (or acting complementarily):
(1) Antibodies aren’t perfect. In fact, a lot of times they suck (bind with low affinity and/or non-specifically). With so many cells and so many antibodies concentrated in solution, it’s very possible off-target binding is happening at least at some low level. Incubating cells with TACs for longer may provide enough time for these non-specific binding events to accumulate on cells and decrease recovery significantly
(2) Many sticky platelets remain in solution (hard to remove with washes b/c of their similar density to PBMCs). It’s possible these platelets aid in cell entrapment by acting as a glue that connects desired, uncoated cells w/ formed lattices or directly w/ the inner walls of the tube.
(3) Settling. With longer incubation times, large cross-linked lattices in solution may naturally settle to the bottom of the tube - in the process, entrapping uncoated (desired) cells with them.
(4) Volume. If you don’t dilute up to the recommended volumes, extra-concentrated solutions of cells+TACs+particles may lead to quicker nucleation and aggregation kinetics
(5) Temperature. If you don’t do everything at room temperature but, for example, use buffer that’s been on ice (4C) this may discourage antibody binding and affect the kinetics/efficiency at which cells are “pulled” out of solution by the magnet
With all this now in mind, let’s proceed to isolation:
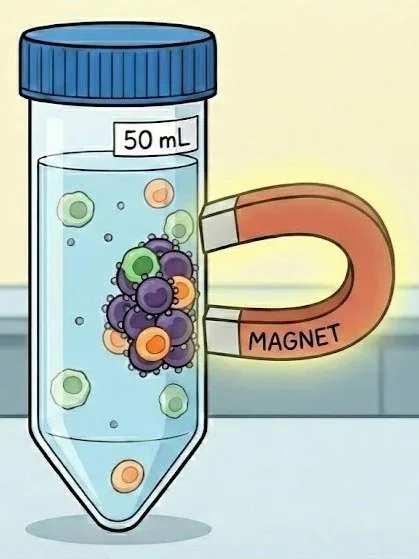
Generated (after many prompts...) w/ Nano banana. I wish it could output figures at higher resolution. Clearly cells not drawn to scale but hopefully you get a sense of how pulling out labeled cells can entrap unlabeled cells
Step 4A: Isolate CD14+ monocytes
These cells are more fragile than lymphocytes so we’re going to process them first, setting the washed PBMCs intended for T cell isolation (“T fraction”) aside for now, maintaining at room temp. in the biosafety cabinet
Note that the two different isolation kits have slightly different protocols!
1. Retrieve EasySep kit from 4C. If time permits, allow 20 min for kit to warm to room temp.
Pre-warming is not all that important here b/c you’re transferring a small vol. of cold liquid into a much large, volume of suspended cells at room temp.
2. Add 1 mL of isolation cocktail (containing TACs) to washed PBMCs suspended in 20 mL EasySep buffer at room temp (“M fraction” from last step). Cap tube then invert gently 3x to mix.
The cell concentration should be ~5E7 TNCs/mL for a total of 5E7*20 = 1E9 TNCs input. Slightly lower concentrations and/or total cell input is fine, higher is not
3. Add 1 mL of platelet removal cocktail (not sure what’s in this, platelet-specific TACs?) to sample. Cap tube then invert gently 5x to mix.
The platelet-removal cocktail is optional. With it, your final cell purity will be higher but your overall cell yield might be slightly lower (b/c platelets can bind to monocytes). I always include.
4. Incubate sample at room temp for 5 min exactly. While waiting, vortex tube of magnetic particles thoroughly to get back into homogenous suspension.
5. Immediately, add 1 mL of vortexed magnetic particles to sample. Pipette 3x to mix then cap sample & invert gently 3x to mix.
6. Incubate sample at room temp for 5 min exactly.
7. Immediately, top up sample to 50 mL total volume with room temp. EasySep buffer. Cap and invert gently 3x to mix.
8. Immediately, loosen cap and apply diluted sample to STEMCELL Easy 50 magnet.
9. Incubate sample RT on magnet for 10 min
10. Carefully, keeping tube on magnet, withdraw 45-50 mL of sample (containing unbound cells to keep) without touching sides of tube or causing liquid level to spill over
11. Transfer collected cell suspension to new 50 mL conical tube and label (“purified CD14+ monocytes“)
12. Spin collected monocytes 220xg for 10 min (w/ brake on)
While waiting, retrieve, label, and uncap 11 cryotubes
13. Carefully remove supernatant without disturbing cell pellet then re-suspend cells in 11 mL of cold CryoStor CS10 cryopreservation medium, pipetting gently to mix (working quickly)
Cells don’t like cryopreservation medium (containing 10% DMSO), the quicker you freeze the better.
14. Working quickly, aliquot 1 mL per cryotube (n=10-11 tubes total) then seal tubes, transfer to styrofoam freezing container, and store at -80C
15. With the small amount of cells remaining (suspended in CS10), dilute 10 uL into 990 uL dPBS OR 100 uL into 900 uL dPBS and measure by flow cytometry
Alternatively, you can use cRPMI as the dilution medium to make the cells happier before flowing. They quickly shrivel up in CS10.
Choose your dilution based on how many cells you think you recovered. If the final pellet was large then dilute 0.01x. If it was small, dilute 0.1x.
16. Next day, transfer cells from -80C freezer to long-term liquid nitrogen storage (-150C).
Boiling point of N2 is -196C. We store cells in nitrogen’s vapor phase which, in the cryotank, ranges from -120C to -180C
Step 4B: Isolate CD8+ T cells
Note that the two different isolation kits have slightly different protocols!
1. Retrieve EasySep kit from 4C. If time permits, allow 20 min for kit to warm to room temp.
Pre-warming is not all that important here b/c you’re transferring a small vol. of cold liquid into a much large, volume of suspended cells at room temp.
2. Add 1 mL of isolation cocktail (containing TACs) to washed PBMCs suspended in 20 mL EasySep buffer at room temp (“T fraction” from last step). Cap tube then invert gently 3x to mix.
The cell concentration should be ~5E7 TNCs/mL for a total of 5E7*20 = 1E9 TNCs input. Slightly lower concentrations and/or total cell input is fine, higher is not
3. Incubate sample at room temp for 5 min exactly. While waiting, vortex tube of magnetic particles (“RapidSpheres”) thoroughly to get back into homogenous suspension.
4. Immediately, add 1 mL of vortexed magnetic particles to sample. Pipette 3x to mix then cap sample & invert gently 3x to mix.
5. Immediately, top up sample to 50 mL total volume with room temp. EasySep buffer. Cap and invert gently 3x to mix.
There is no secondary incubation step once the magnetic beads have been added for the T cell isolation kit, hence the name “RapidSpheres”. Idk why they don’t use these same beads for the monocyte kit, possibly because monocytes bind dextran?
6. Immediately, loosen cap and apply diluted sample to STEMCELL Easy 50 magnet.
7. Incubate sample on magnet at room temp. for 10 min (“pulldown #1”)
8. Carefully, keeping tube on magnet, withdraw 45-50 mL of sample (containing unbound cells to keep) without touching sides of tube or causing liquid level to spill over
9. Transfer collected cell suspension to new 50 mL conical tube
10. Loosen cap and incubate sample (in new tube) on magnet at room temp. for 10 min (“pulldown #2”)
11. Carefully, keeping tube on magnet, withdraw 45-50 mL of sample (containing unbound cells to keep) without touching sides of tube or causing liquid level to spill over
12. Transfer collected cell suspension to new 50 mL conical tube and label (“purified CD8+ T cells “)
13. Spin collected T cells 300xg for 10 min (w/ brake on)
While waiting, retrieve, label, and uncap 11 cryotubes
14. Carefully remove supernatant without disturbing cell pellet then re-suspend cells in 11 mL of cold CryoStor CS10 cryopreservation medium, pipetting gently to mix (working quickly)
Cells don’t like cryopreservation medium (containing 10% DMSO), the quicker you freeze the better.
15. Working quickly, aliquot 1 mL per cryotube (n=10-11 tubes total) then seal tubes, transfer to styrofoam freezing container, and store at -80C
16. With the small amount of cells remaining (suspended in CS10), dilute 10 uL into 990 uL dPBS OR 100 uL into 900 uL dPBS and measure by flow cytometry
Alternatively, you can use cRPMI as the dilution medium to make the cells happier before flowing. They quickly start to shrivel up in CS10 (within 5-15 min)
Choose your dilution based on how many cells you think you recovered. If the final pellet was large then dilute 0.01x. If it was small, dilute 0.1x.
17. Next day, transfer cells from -80C freezer to long-term liquid nitrogen storage (-150C).
Final results
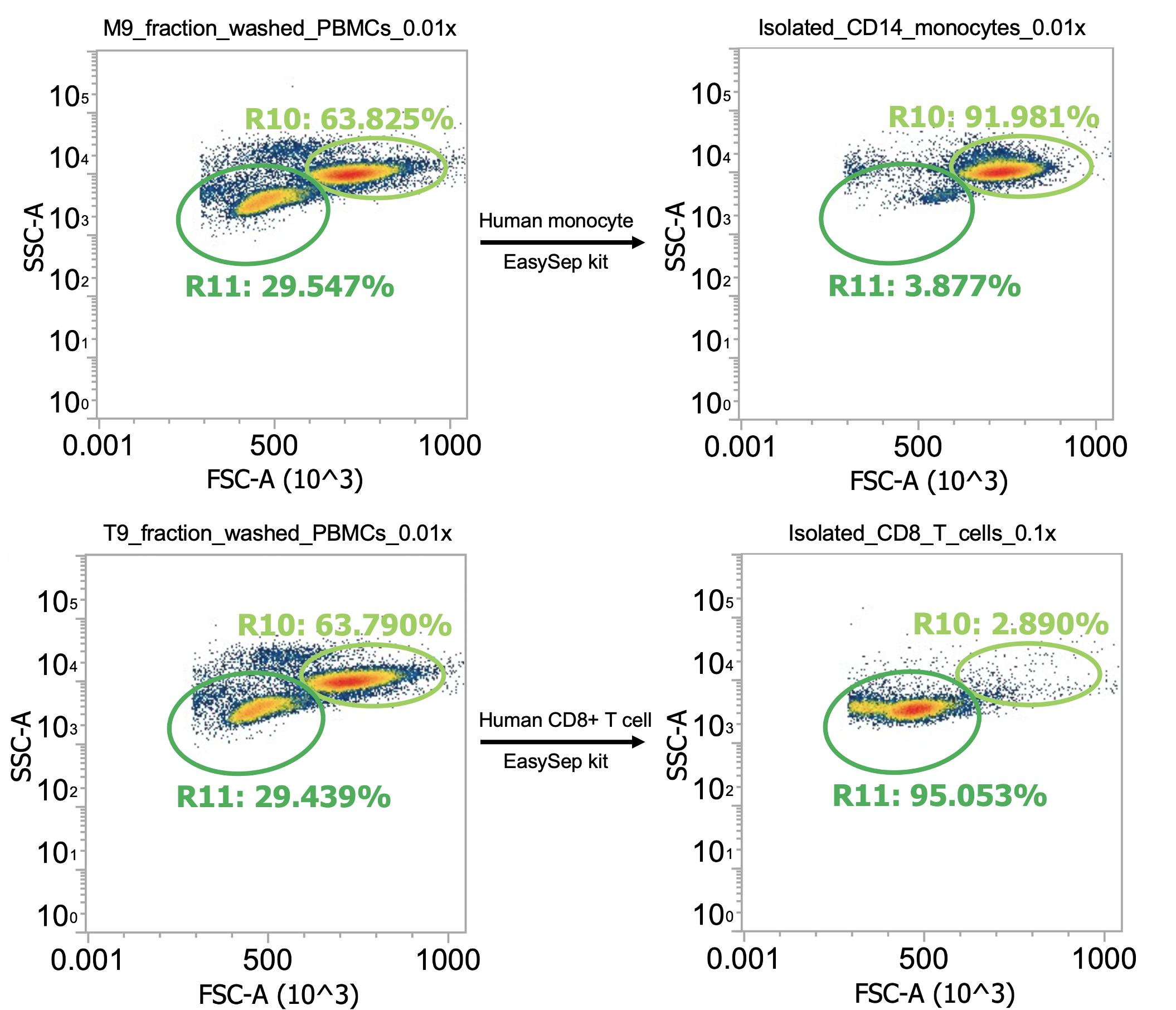
Figures show gated cells (ignoring debris) which represented > 80% of total events in final samples. In bottom right panel, you can see the CD8s starting to die by the end of the isolation w/ events creeping down in FSC-A and up in SSC-A (cells shriveling).
Hopefully, all of this has, in some way, helped you obtain higher & purer yields from your primary immune cell isolation work!
I’ll post updates as I process more leukopaks and learn more.
Monocyte yield (adhering to recommended EasySep kit incubation times):
Input = 3.8e7 TNCs/mL * 20 mL * 64% monocytes * 90% CD14+ (est.) = 440e6 CD14+ monocytes
Output = 2.9e7 cells/mL * 11 mL * 92% monocytes = 290e6 CD14+ monocytes (66% yield)
Immunostaining isolated cells w/ fluorescent mouse anti-human CD14 antibodies confirms >90% of cells are the desired CD14+ monocytes (data not shown)
Staining w/ DAPI confirms cells are > 85% healthy (data not shown)
T cell yield (not adhering to recommended EasySep kit incubation times; incubated longer)
Input = 3.8e7 TNCs/mL * 20 mL * 30% lymphocytes * 30% CD8+ (est.) ~= 70 million CD8+ T cells
Output = 1.4e6 cells/mL * 11 mL * 95% T cells = 15 million CD8+ T cells (20% yield… low)
Immunostaining isolated cells w/ fluorescent mouse anti-human CD3, CD8, and CD4 antibodies confirms >95% of cells are the desired CD3+CD8+ T cells (data not shown)
Staining with DAPI reveals cells are ~67% healthy (33% of cells are DAPI+, the shifting/dying group of cells stemming off the primary population in the rightmost figure)
Note: This was an unusual leukopak donation (or lymphoprep separation) wherein monocytes outnumbered lymphocytes 2:1 in the isolated PBMCs. Usually, it’s the other way around.
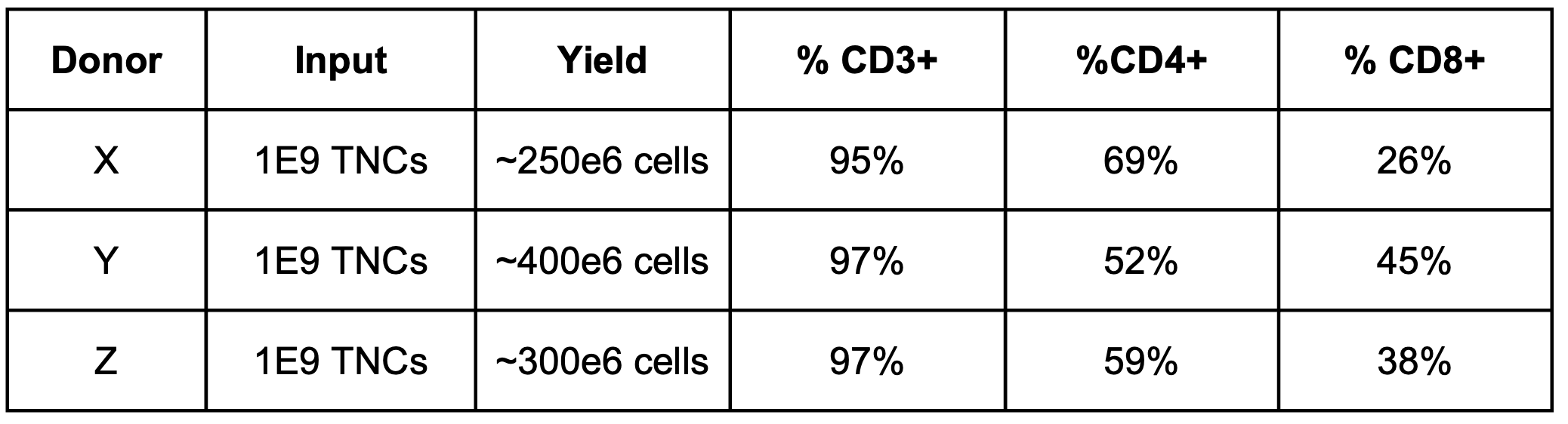
To further test my theory regarding incubation times, I isolated CD3+ T cells from 3 additional leukopaks using STEMCELL’s EasySep Human T cell isolation kit (cat # 17951) that purifies both CD4+ and CD8+ T cells, washing cells twice at 300g for 10 min then following the kit protocol strictly, with the following cell yields (and purity, measured by extracellular immunofluorescent staining):
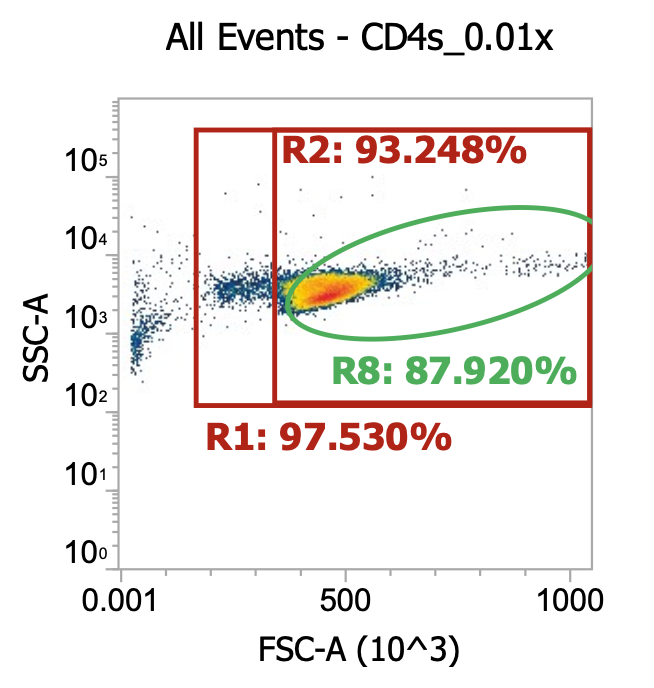
Isolating (autologous) CD4s, CD8s, and CD14s in parallel
Updates! Indeed, to obtain high yields & purities w/ STEMCELL’s EasySep kits, it appears you must strictly follow the recommended antibody & magnet incubation times, as outlined above. To prove it, here are the results of my latest isolation (Donor 10), where I purified CD4s, CD8s, and CD14s separately, but in parallel, from a single ¼ leukopak in (a busy) 3-4 hours.
I got this ¼ leukopak from STEMCELL for free as part of a March promotion (after ordering $10k worth of kits…)
Typically, suppliers guarantee >2.5 TNCs from a ¼ leukopak. This one arrived with a supplier estimated 6.5E9 TNCs (in 84 mL) which is very good… let’s see if it’s accurate
Note: I asked if they could ship the leukopak at ambient temp. since my most recent experience is that monocytes hold better at room temp. but they ignored and shipped at 4C (oh well). Altogether, shipping, receipt, and processing was done within ~24h of collection which is ideal.
Instead of providing the step-by-step details directly in the webpage, here’s a PDF:
For the remainder of the post I’ll just share the key #s and flow plots
Overall, my approach was to isolate cells in the following order:
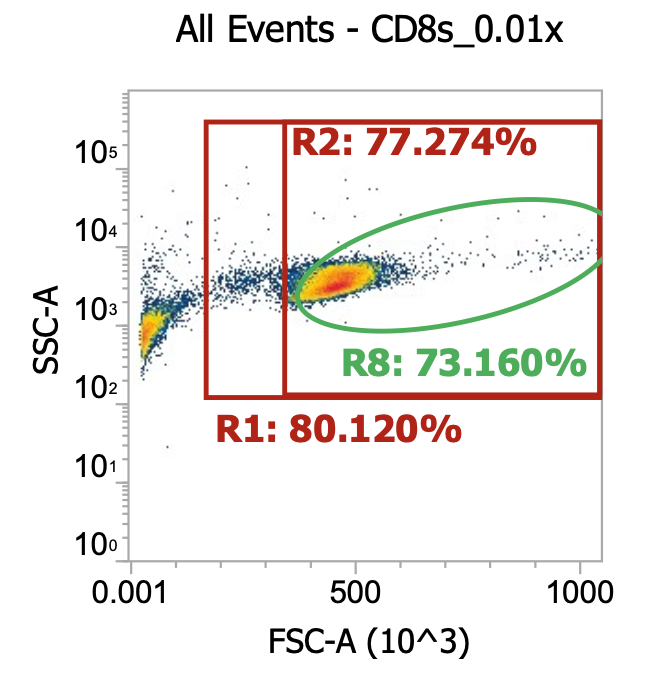
CD4s (after 2x simple washes of crude leukapharesis product)
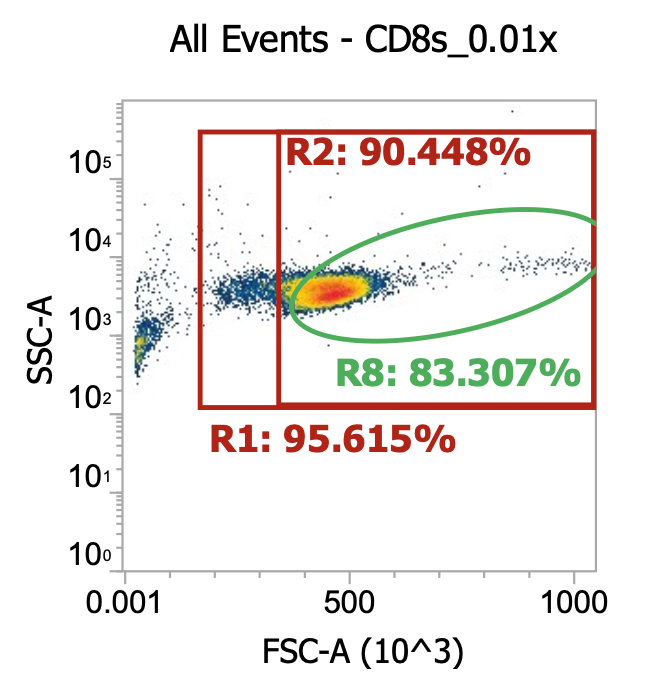
CD8s (after PBMC isolation followed by 1x gentle wash)
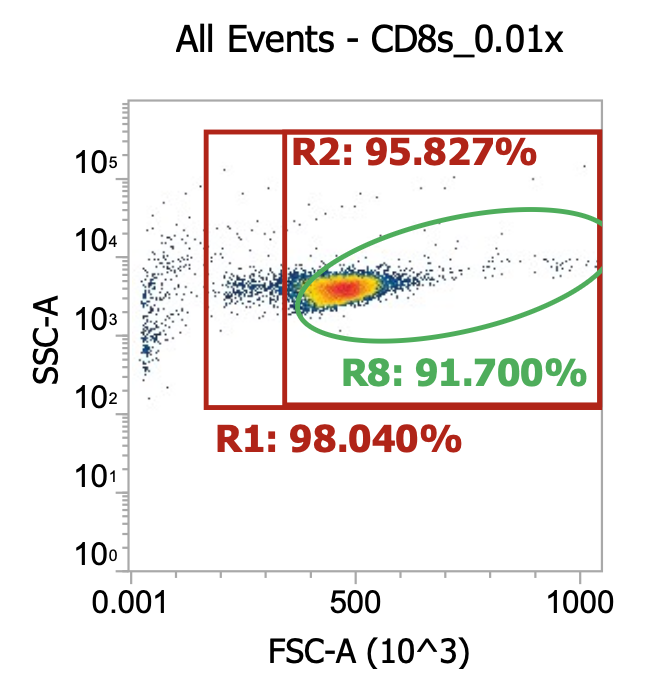
CD14s (after PBMC isolation followed by 2x gentle washes)
The different processing methods were to test out how much the leukapharesis product really needs to be cleaned up before inputting into the EasySep kits. It also let me temporally separate the isolation steps b/c I only have one EasySep magnet. Also, I care about the monocytes the most and CD8s more than CD4s so the more important the cell type the more I washed the input prior to isolating.
I started by splitting the sample into two groups:
2/3 of the (DNase I treated) leukapheresis product (60 mL, allegedly ~4.5E9 TNCs) went into PBMC isolation
1/3 of the (DNase I treated) leukapheresis product (24 mL, allegedly ~2E9 TNCs) went into CD4 isolation
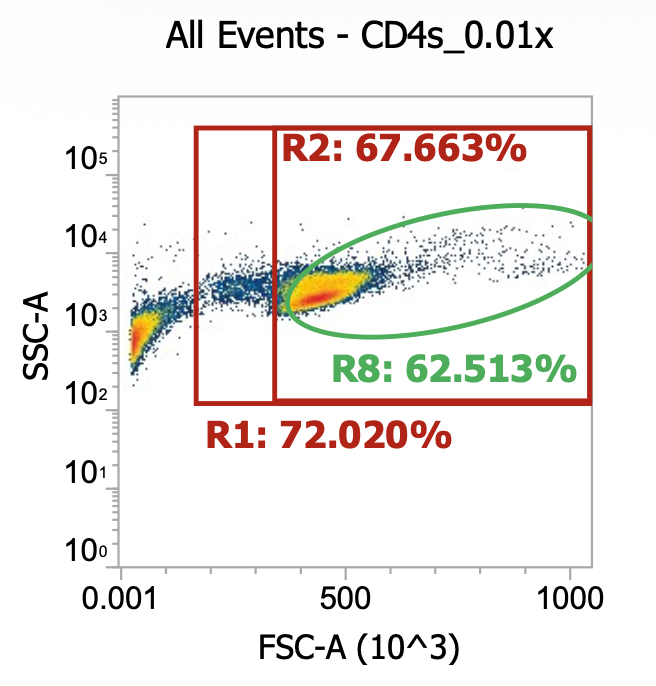
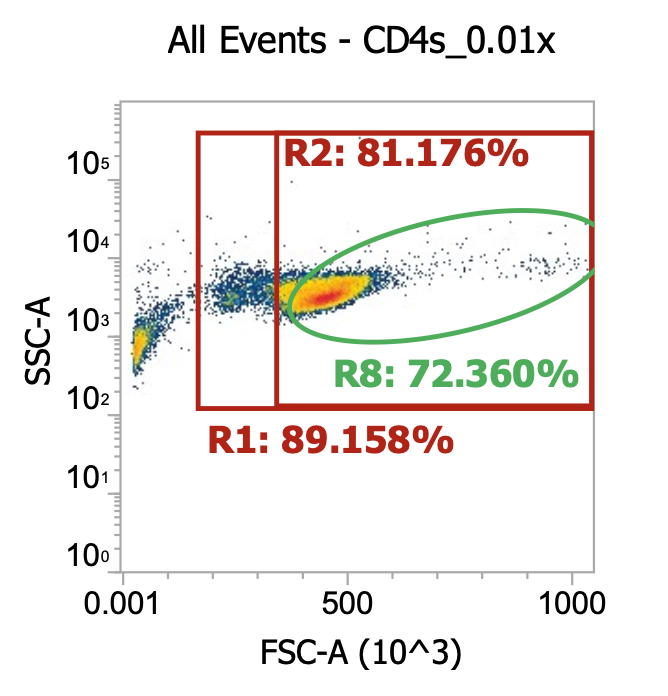
For the cells intended for CD4 isolation, I split the volume across 2×50 mL tubes (12 mL per tube) and diluted up to 50 mL with EasySep buffer (dPBS + 2% FBS + 1 mM EDTA) then spun 300xg, 10 min w/ brake on. Then, I resuspended each cell pellet in 50 mL EasySep buffer and spun once more. Lastly, I resuspended each pellet in 22 mL buffer and combined for a total of ~45 mL of 2x washed cells. I took a 10 uL of this final suspension and diluted into 990 uL of dPBS then ran 100 uL of this 0.01x dilution through our flow cytometer.
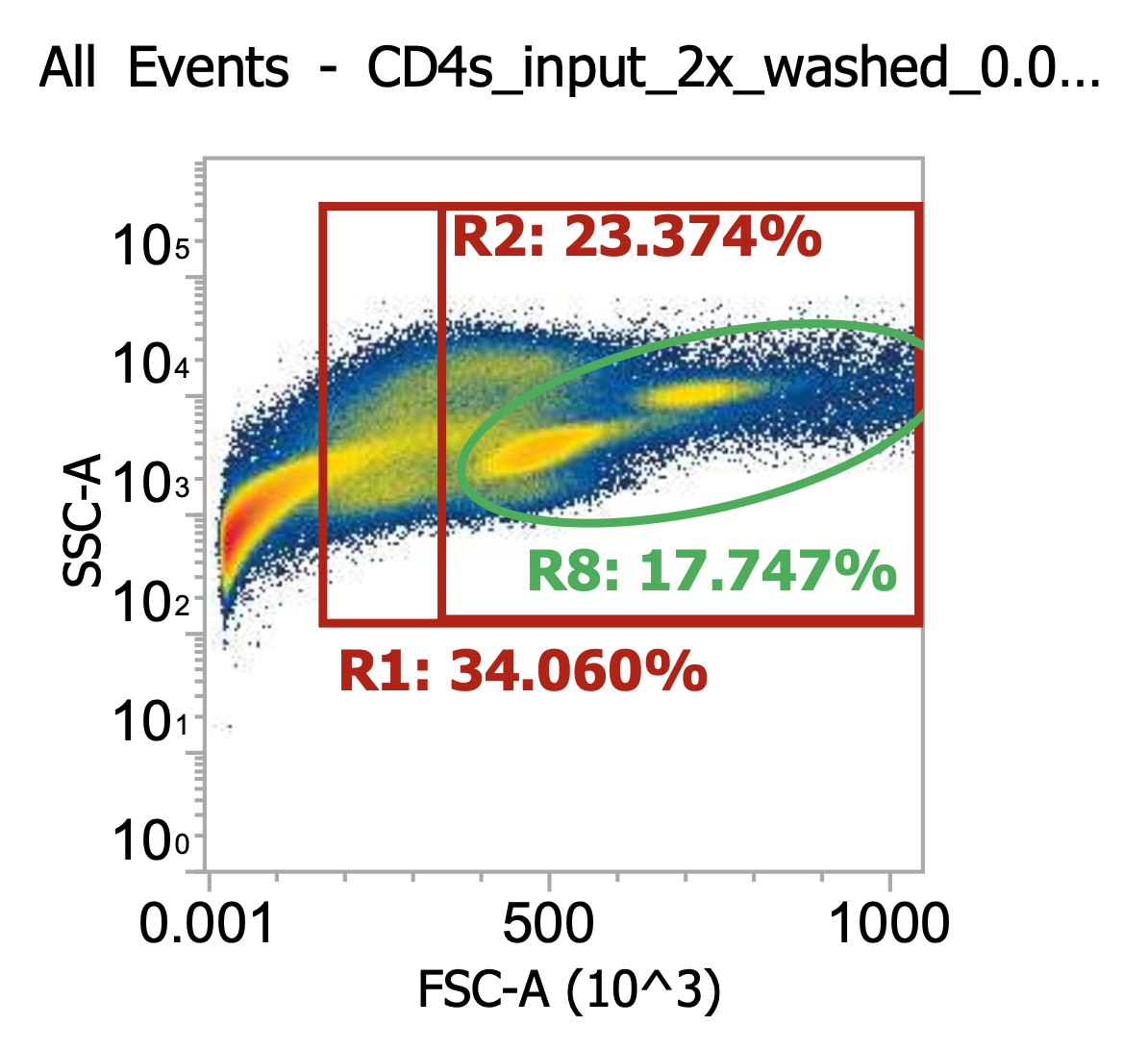
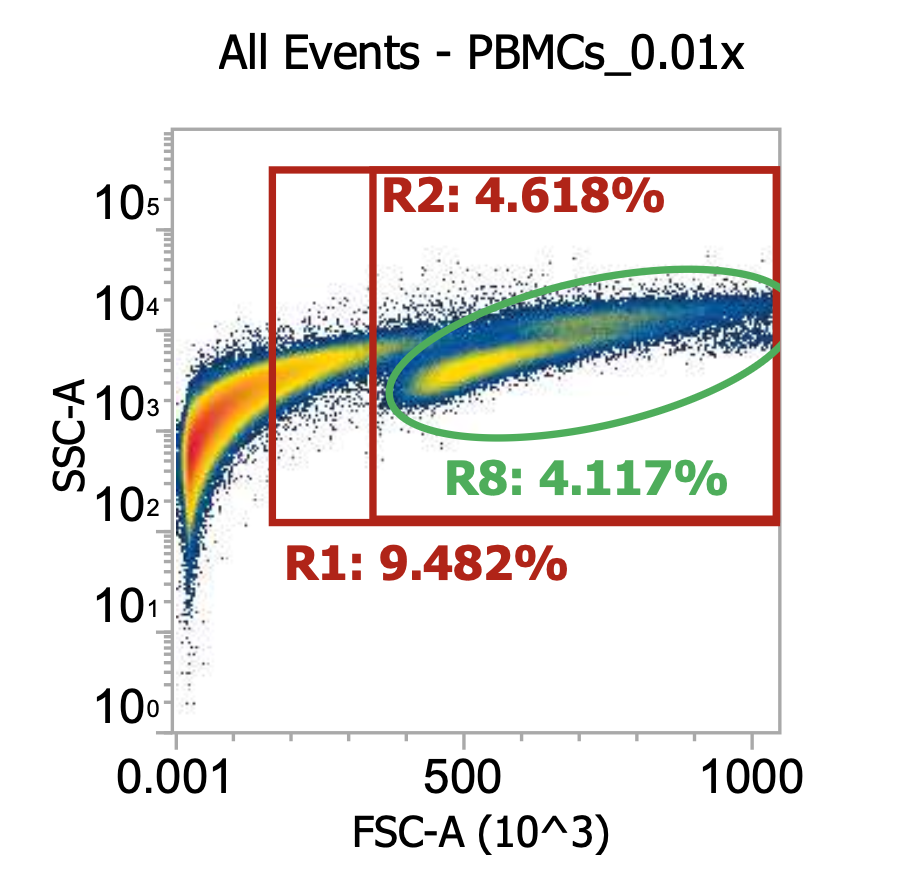
As you can see, even after a couple washes, the leukapheresis product is still quite messy. The red R1 gate captures all TNCs once the sample is cleaned up some more (see below) but for now there are lots of platelets, RBCs, and general cell/blood debris spilling into it.
The green R8 gate loosely captures both lymphocytes (lower FSC/SSC) and monocytes (higher FSC/SSC). Outside of the R8 gate but within the TNC (R1) gate you can also kind of make out a population of neutrophils w/ high SSC.
R8 gate count = 100,000 events * (1000/100 uL) * 100 = 100e6 cells/mL * 45 mL = 4.5E9 TNCs / mL (est.)
This is a very rough approximation b/c the sample is so messy. For now, let’s lean on STEMCELL’s estimate a bit and assume we actually have ~2E9 TNCs in the washed sample (i.e. negligible losses from centrifuging @ 300xg).
Assuming that to be the case, let’s then choose to input 30 mL out of the 45 mL total into the CD4 EasySep kit which accepts up to 1E9 TNCs. Yes, that means that we’d be slightly over the limit, but it’s close enough - that’s not the critical part of the protocol (although I’m sure STEMCELL would disagree).
Proceed to isolate CD4s as detailed in the PDF (essentially, just following STEMCELL’s protocol strictly). Once isolated, centrifuge cells once more 300xg, 10 min w/ brake on and then re-suspend final pellet in 11 mL of CS10 freezing medium, aliquot to labeled cryotubes, and store at -80C. With the small amount of residual volume leftover in the tube, dilute 10 uL into 990 uL dPBS and flow 200 uL of this 0.01x dilution on the flow cytometer.
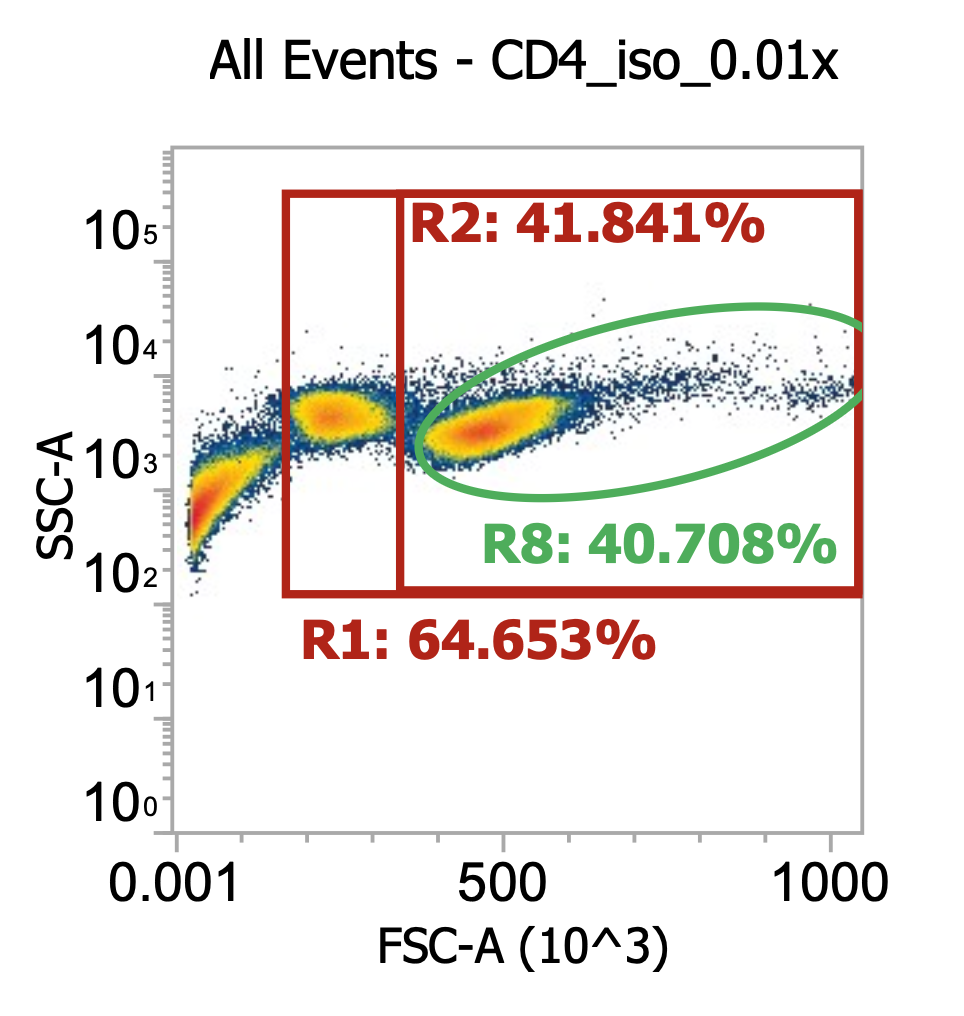
Three distinct populations in the final purified CD4s. The first is leftover debris to the far left representing ~35% of the total events. We can live w/ that. The second is the cells gated by R8, our healthy TNCs which are heavily enriched w/ lymphocytes and depleted of monocytes
The third (middle) population I believe represents dying lymphocytes. They start to shrivel up and decrease by FSC-A while slightly increasing in SSC-A. If I were to stain this sample w/ DAPI, the cells in this population would light up indicating porous (dying) membranes.
I’m surprised to see so many dying lymphocytes (40% of isolated TNCs), especially since these are the first ones we’ve isolated from the leukopak, within approx. 1 hours of receiving the shipment. It’s not a huge deal though b/c we still have lots of healthy cells:
R8/R11 gate count = 37,500 events * (1000/200uL) * 100 * 11 mL = 206e6 healthy CD4s (!)
(This represents 50-75% of the theoretical CD4 yield from 1E9 leukopak-derived TNCs)
Now, I’m assuming all/most of the isolated cells are CD3+CD4+ T cells however I will confirm this by culturing and staining the saved cells in the coming weeks. For now, onto the next kit!
While CD4 isolation was taking place, I had already begun isolating PBMCs from the other 60 mL of leukapheresis product. For this, I split the designated sample fraction into 6×10 mL aliquots and diluted each w/ 20 mL EasySep buffer, mixing by inversion. I then carefully layered these 6×30mL samples on top of 6×15 mL aliquots of STEMCELL’s lymphoprep solution careful to maintain phase separation. I then spun these 6×45 mL samples 800xg for 30 min w/ brake off so it actually took ~40 min. Afterwards, I pooled the buffy coats from each tube (~ 8 mL / tube) together for a total of 45 mL of isolated PBMCs. I then diluted a 10 uL aliquot into 990 uL dPBS and ran 100 uL of this 0.01x dilution on our flow cytometer.
Still lots of spillover of platelets / debris into the TNC gates (R1/R2), however, neutrophils (and RBCs) now depleted by nature of separating via density gradient
We’ve actually enriched for platelets here since they have similar density and separate w/ the PBMCs
R8 gate count = 76,600 events * (1000/100uL) * 100 = 77e6 TNCs/mL …*45 mL = 3.5E9 TNCs/PBMCs total (>80% recovery)
Next, I split the PBMCs as 2×22 mL aliquots, diluted up to 50 mL w/ EasySep buffer, then spun 250xg for 10 min w/ brake off (takes 15-20 min total). I then discarded the (very cloudy) sup. and resuspended each cell pellet in 30 mL EasySep buffer. Replicate samples are now considered 1x (gently) washed. Designated one replicate for CD8 isolation and the other for CD14 isolation. For the latter (monocyte fraction), I again diluted up to 50 mL w/ buffer then spun again at 250xg for 10 min w/ brake off. Meanwhile, for the former (CD8 fraction), I diluted a 10 uL aliquot into 990 uL dPBS and ran 100 uL of this 0.01x dilution on the flow cytometer.
One gentle wash enriches TNCs from ~4% to ~14% of the total events (>3x). Not bad.
R8 gate count = 44,000 TNCs * (1000/100uL) *100 = 43.5e6 cells/ml * 30mL = 1.32E9 TNCs
Implying 2×1.32E9 = 2.65E9 TNCs recovered from 3.5E9 input after 1x gentle wash
I.e. 75% recovery per gentle wash step (not great, not terrible)
Interesting to look at the cell type ratio here as it matches general expectations:
Lymphocytes: 56/85 = 66% of TNCs
Monocytes: 29/85 = 33% of TNCs
Proceeded to input 25 of 30 mL of 1x gently washed PBMCs into the EasySep CD8 isolation kit (see PDF for details). I then spun down the isolated cells 250xg for 10 min w/ brake on to pellet then resuspended in 11 mL of CS10, aliquoted to cryotubes, and saved at -80C. Lastly, I diluted 10 uL of residual sample into 990 uL of dPBS and ran 200 uL of this 0.01x dilution on the flow cytometer.
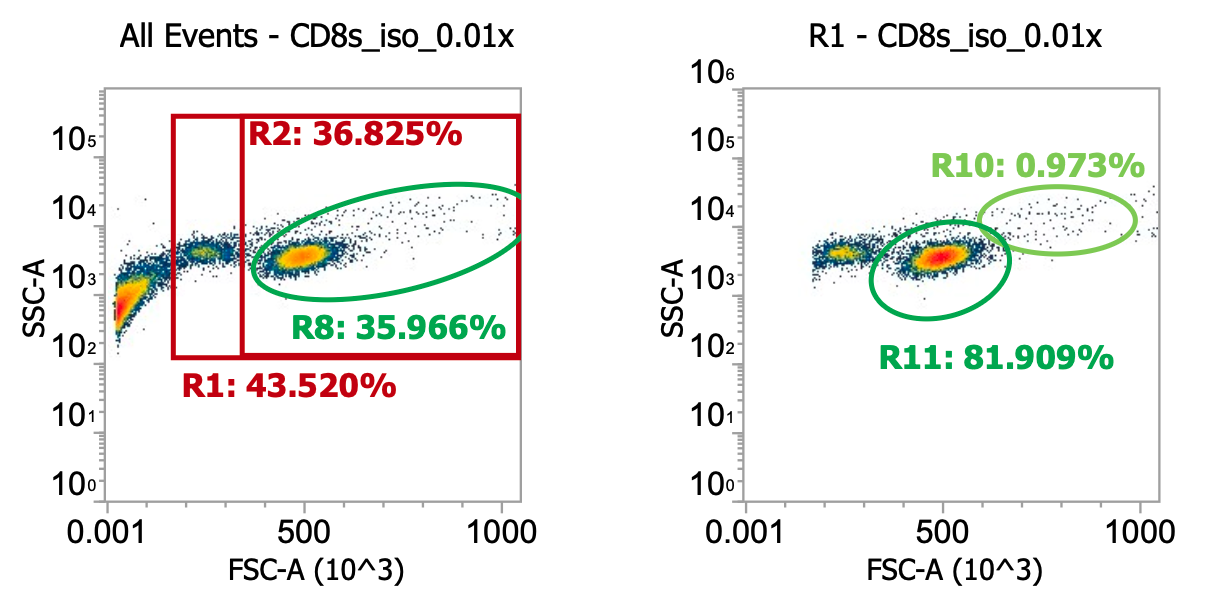
R8 gate count = 9,589 events * (1000/200uL) * 100 * 11mL = 52.7e6 healthy CD8s
Estimated 25-50% recovery of total theoretical CD8s in 1E9 TNCs from leukopak
Clear monocyte depletion (<1% of TNC events)
Platelets / debris still account for ~60% of total events but this is fine, doesn’t really affect downstream culturing and will naturally deplete over time / with washes
~15% of isolated TNCs appear to be dying lymphocytes by FSC-A/SSC-A. Not great, not terrible. Will know more after thawing, culturing, and immunostaining.
Sidenote: I told you the R1 gate would eventually be a good TNC discriminant
So far so decent! 200e6 CD4s and 50e6 CD8s. Now onto the monocytes…
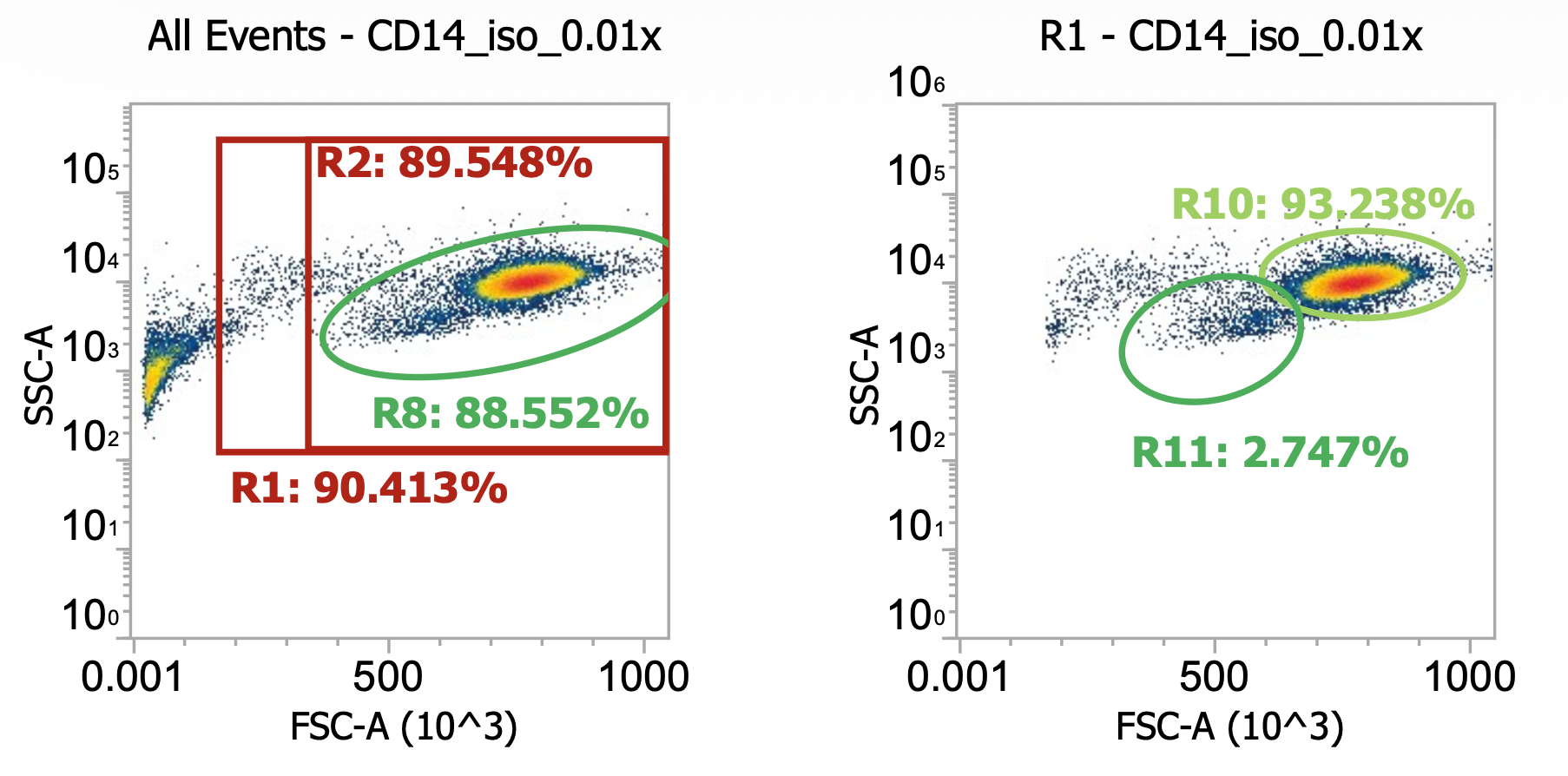
Re-suspend the now 2x gently washed PBMCs designated for CD14 isolation in 20 mL EasySep buffer and dilute 10 uL aliquot in 990 uL dPBS then run 100 uL of this 0.01x dilution on flow cytometer.
So we went from 4% TNCs in buffy coat (crude PBMCs) to 14% after 1x gentle wash to, now, 22%
Again, the split of lymphocytes-to-monocytes is notable as it matches expectation
R11 gate) 56/(56+29) = 66% lymphocytes
R10 gate) 29/(56+29) = 33% monocytes
Note how the ratio didn’t change after an additional wash indicating a sufficient centrifugation speed
R8 gate count = 55,000 events * (1000/100uL) * 100 = 55e6 cells/mL * 20 mL = 1.1E9 TNCs total
Perfect! Input all 20 mL of cell suspension into CD14+ EasySep isolation kit (see PDF for details)
Spin down final, isolated CD14s by 220xg for 10 min w/ brake on then resuspend in 11.5 mL of CS10, dispense to cryotubes, and save at -80C. Dilute 10 uL of residual volume (leftover sample) in 990 uL dPBS and run 200 uL of this 0.01x dilution on flow cytometer
R10 gate count = 35,000 * (1000/200uL) * 100 × 11 mL = 192e6 healthy CD14s
Extremely pure, representing >90% of total events in sample and >93% of total TNCs detected
A theoretical recovery of 60-80% based on expectation from 1E9 leukapharesed TNCs
I think the fact that the EasySep kit for CD14+ monocyte isolation includes a secondary “platelet removal cocktail” (antibody mix) really helps w/ final sample purity compared to the CD4/CD8 kits
Hopefully now I’ve convinced you that my gates successfully partition all TNCs (R1), healthy TNCs (R2/R8), lymphocytes (R11) and monocytes (R10). Key to the success of flowing/quantifying all these samples in real time is diluting 1:100 as to not oversaturate the laser and/or clog the lines.
Also helpful is careful prior tuning of cytometer voltages & plotting FSC-A/SSC-A by linear/log scale.
Summary
And there you have it! Tens to hundred of millions of isolated immune cells using immunomagnetic separation. All completed within 4 hours of receiving a ¼ leukopak.
We allegedly started w/ ~6.5e9 TNCs which, in retrospect, appears pretty accurate
We input 70% of the cells (4.5E9 TNCs) into PBMC isolation via density separation and recovered 3.5E9 TNCs in the buffy coat (~80%)
Gently washing the PBMCs enriched for TNCs (depleted platelets) but also lost some cells (25% per wash). It’s a careful balance b/c the densities are so similar.
3.5E9 / 2 = 1.75E9 TNCs * 0.75 ~= 1.3E9 TNCs retained for CD8 isolation (after 1 wash)
3.5E9 / 2 = 1.75E9 TNCs * 0.75² ~= 1.0E9 TNCs retained for CD14 isolation (after 2 washes)
| Isolation kit | Cell source | Preprocessing method | Input TNCs | Input composition | TNCs recovered | % healthy | Est. total recovery |
|---|---|---|---|---|---|---|---|
| EasySep Human CD4+ T cell isolation (negative selection) |
Leukopak | 2x washes (300g, 10 min) |
~1.5E9 | 33% monocytes / 67% lymphocytes |
330e6 | 62% | 50-75% |
| EasySep Human CD8+ T cell isolation (negative selection) |
Leukopak | PBMCs, 1x wash (250g, 10 min) |
1E9 | 33% monocytes / 67% lymphocytes |
65e6 | 82% | 30-50% |
| EasySep Human monocyte isolation (negative selection) |
Leukopak | PBMCs, 2x wash (250g, 10 min) |
1E9 | 33% monocytes / 67% lymphocytes |
205e6 | 93% | 60-80% |
All cells sourced from the same ¼ leukopak. Estimated recovery assumes a "monocyte" population that is 80-90% CD14+CD16- and a lymphocyte population containing 70% T cells (i.e. 30% B cells & NK cells) with a CD4:CD8 ratio of 2:1By FSC/SSC it appears the isolated cells are the correct immune cell type but I’ll share the staining data when I have it!
Had some time this week so thawed 1 vial of each isolated cell type from Donor 10 and immediately stained using custom monocyte and T cell antibody panels. Histograms show fluorescence of singlet (green) cell population, gated as indicated by the far left dot plots of FSC-A vs. SSC-A, and compared to unstained cells (“control”).
Isolated monocytes are 93% CD14+ and 97% viable whereas isolated CD4s are 96% CD3+CD4+ and 92% viable and isolated CD8s are 85-90% CD3+CD8+ and 95% viable. Overall, not bad!
For the dot plots, datapoint opacity is scaled to 0.1 to better visualize unique clusters of cells. For the histograms, both bins (n=100) & KDE curves are included to visualize the distributions of fluorescent values.
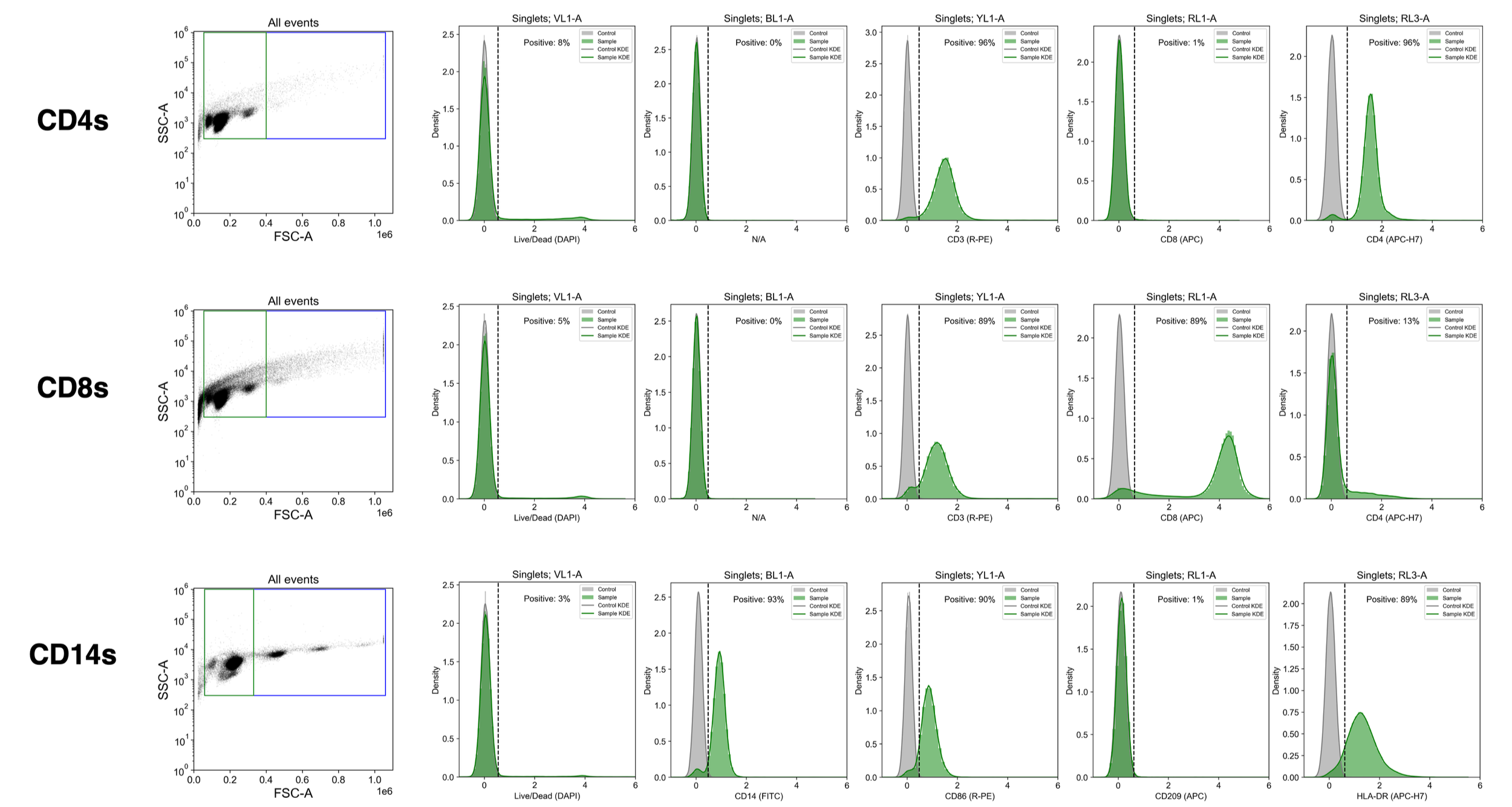
Data gathered on Attune Nxt flow cytometer and plotted in Python using the FlowCytometryTools package. Raw fluorescence values were compensated for spillover and arcsinh transformed (with cofactor=300) for visualization. Gates set manually. Event counts & DAPI staining confirm that cells froze down well in CS10 (stored at -80C for 72h then transferred to vapor phase of liquid nitrogen tank). Per thawed (1 mL) vial, I recovered:
27e6 CD3+CD4+ T cells (>90% viable)
6e6 CD3+CD8+ T cells (>90% viable)
17e6 CD14+ monocytes (>95% viable)
That concludes this protocol! Thanks for reading all the way through, hope all these details helped you in some way :)
Btw, I often share my results w/ STEMCELL, their representatives are very nice, and they provided these estimates of leukopak composition for my target cell types. Great sample sizes!
Numbers match my observations, monocyte content can be highly variable and typical CD4:CD8 ratio is 2:1

Final update!
I isolated autologous immune cells from the leukapharesis product of 3 additional healthy human donors following the protocol/details provided above with the following yields:
| Donor | Format | Total TNCs | Collection date | Isolation date | Total purified, healthy CD4s* |
Total purified, healthy CD8s* |
Total purified, healthy CD14s* |
Total purified, healthy autologous immune cells |
|---|---|---|---|---|---|---|---|---|
| 11 | 1/4 leukopak | 6E9 | 6/11/26 | 6/12/26 | 180e6 | 90e6 | 236e6 | 506e6 |
| 12 | 1/4 leukopak | 4E9 | 6/17/26 | 6/18/26 | 210e6 | 120e6 | 132e6 | 462e6 |
| 13 | 1/4 leukopak | 4E9 | 6/25/26 | 6/26/26 | 203e6 | 93e6 | 120e6 | 416e6 |
* presumably (pre-staining) as estimated by FSC/SSCSo 400e6 - 500e6 total isolated autologous immune cells from 1/4 leukopak:
1/4 leukopak ~= $2,000
3x EasySep kits ~= $3,000
CS10 freezing medium & EasySep buffer = negligible
$600 for enough media (100 mL) & buffer (1L) to isolate cells from 3 donors (i.e. $200 per donor)
$5,000 / 500e6 = $10 / 1e6 human immune cells i.e. $100 / 10e6 immune cells
Compared to ~$1,000 / 10e6 human immune cells if purchased pre-purified (!!!)
In other words, a 90% discount for 3-4 hours of your time!
Donor 11
Donor 12
Donor 13