Packaging lentivirus
Lentivirus is a genus of retroviruses (viruses that insert a DNA copy of their RNA genome into the genome of a host cell)
that packages 2 copies of a positive sense, single-strand (ss)RNA genome into a capsid within virions that are ~100 nm in diameter (~1/100th the diameter of human immune cells). Also known as “slow viruses”, lentivirus cause chronic and deadly diseases in certain mammals including humans.
The genus contains 10 known species which, altogether, have been documented to infect apes, humans, cows, horses, sheep, goats, and cats. Some of these species and their primary hosts are…
Lentivirus humimdef1, Human immunodeficiency virus 1 (“HIV-1”, evolved from SIVcpz in chimpanzees)
Lentivirus humimdef2, Human immunodeficiency virus 2 (“HIV-2”, evolved from SIVsm in sooty mangabees)
Lentivirus simimdef, Simian immunodeficiency virus (“SIV”, note: “simian” = relating to apes & monkeys)
Lentivirus bovimdef, Bovine immunodeficiency virus ('“bovine” = relating to cattle)
Lentivirus equinfane, Equine infectious anemia virus (“equine” = relating to horses)
Lentivirus felimdef, Feline immunodeficiency virus (“feline” = relating to cats)
and others…
Components of HIV-1 / HIV-2 genome (~10kb)
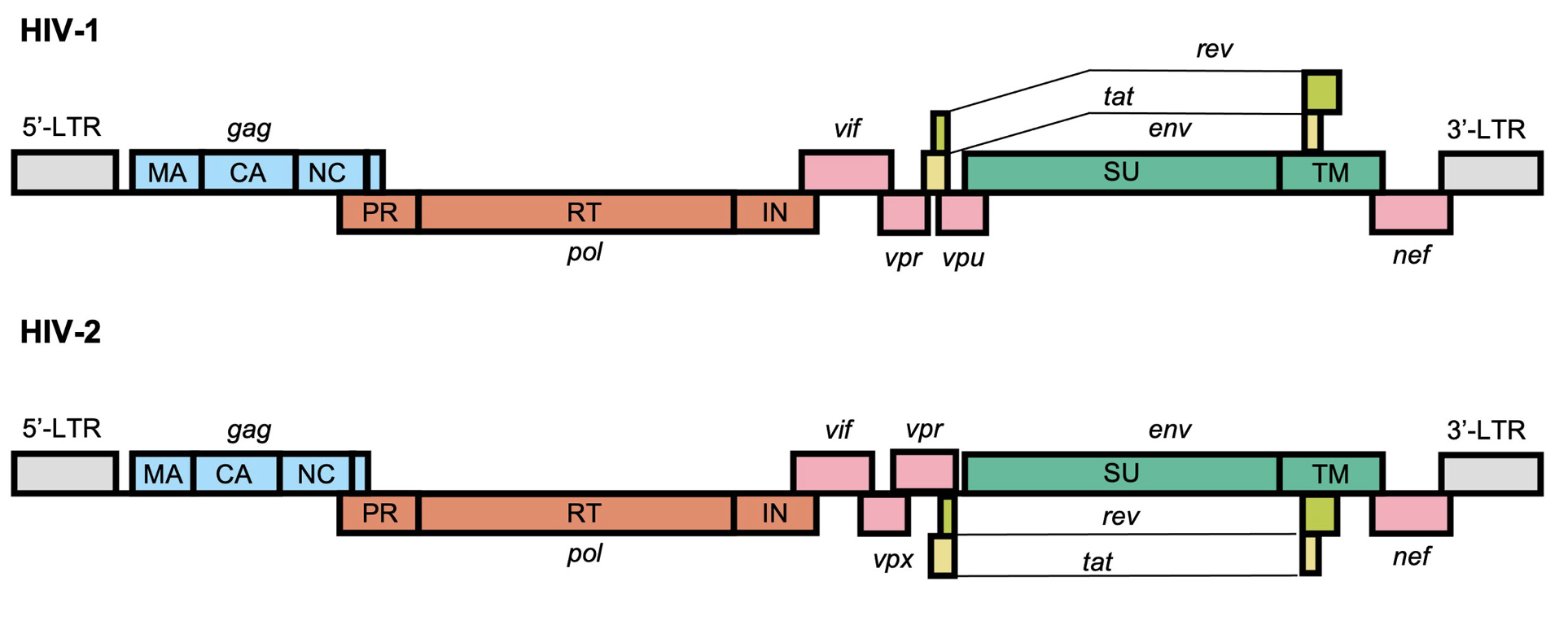
From https://www.frontiersin.org/journals/virology/articles/10.3389/fviro.2022.872599/full
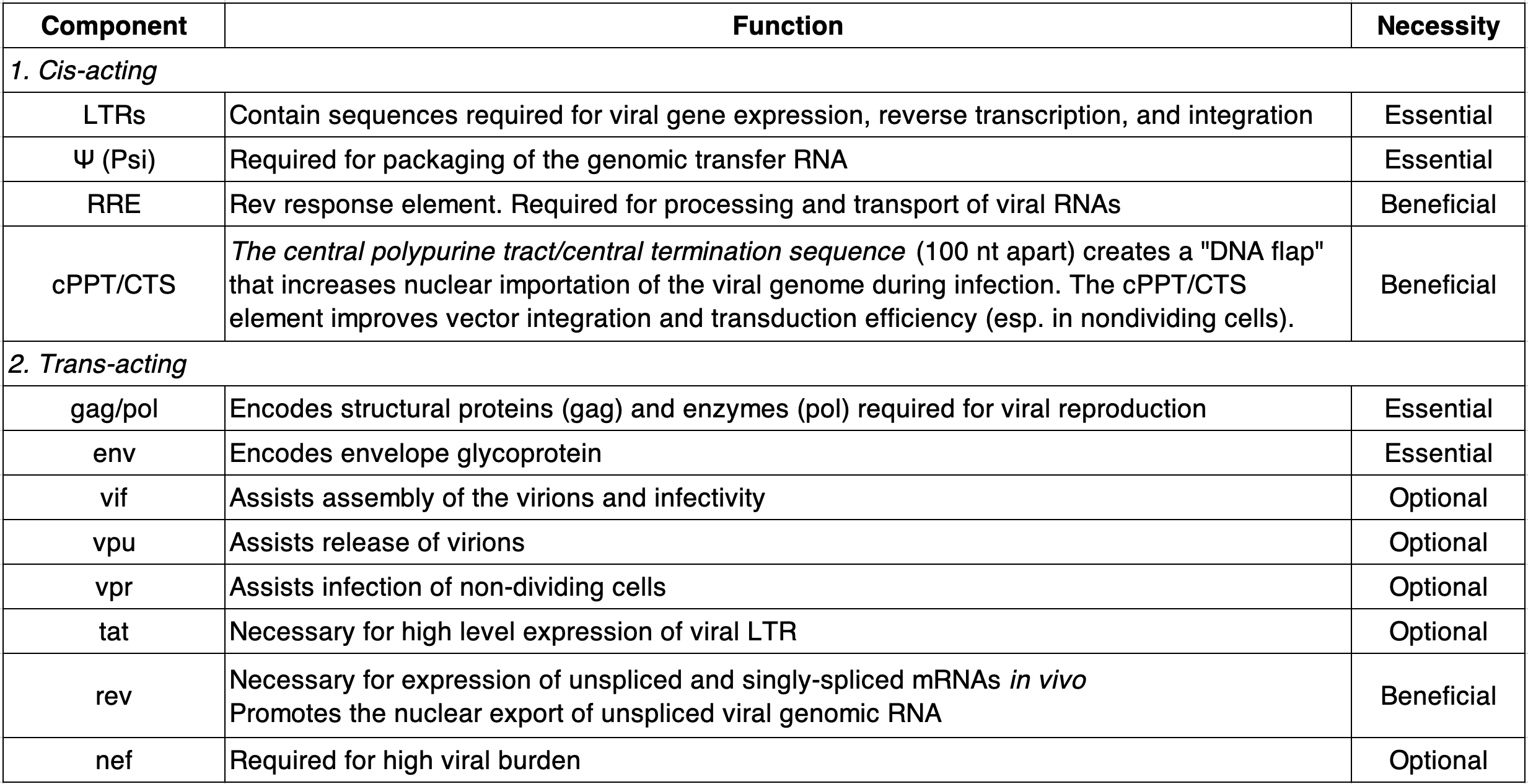
Table largely reconstructed from https://info.abmgood.com/lentivirus-system-introduction
What makes lentivirus so dangerous in humans? It targets immune cells (specifically, CD4+ helper T cells) which reduces their ability to function as well as their overall abundance over time leading to “acquired immunodeficiency disease” (AIDS) in which the the body can’t fight off infection, or suppress neoplastic cells, due to limited immune surveillance and response.
Even though HIV is not (yet) curable (b/c the virus integrates in the genome of host cells making it very tricky to selectively target & eliminate), antiretroviral therapies (ART) strongly suppress it, allowing people with HIV to live long and healthy lives while simultaneously reducing the risk of transmission.
Perhaps equally as amazing as ART, scientists have co-opted lentiviruses for research. Because of its relatively large genome size and ability to infect both dividing and nondividing human cells, (attenuated) lentivirus is a great tool for delivering up to ~10kb of exogenous genetic material into many cells types of interest. Even more useful, it doesn’t just introduce genetic material into cells but stably integrates it into the host genome meaning we can “permanently” modify the genome of cells and study how their behavior/functions then change. This technique was developed by identifying & isolating the genetic components of lentivirus that are essential for producing infective virions & integrative RNA onto plasmids while omitting or modifying components that enable successive viral replication (see table, above).
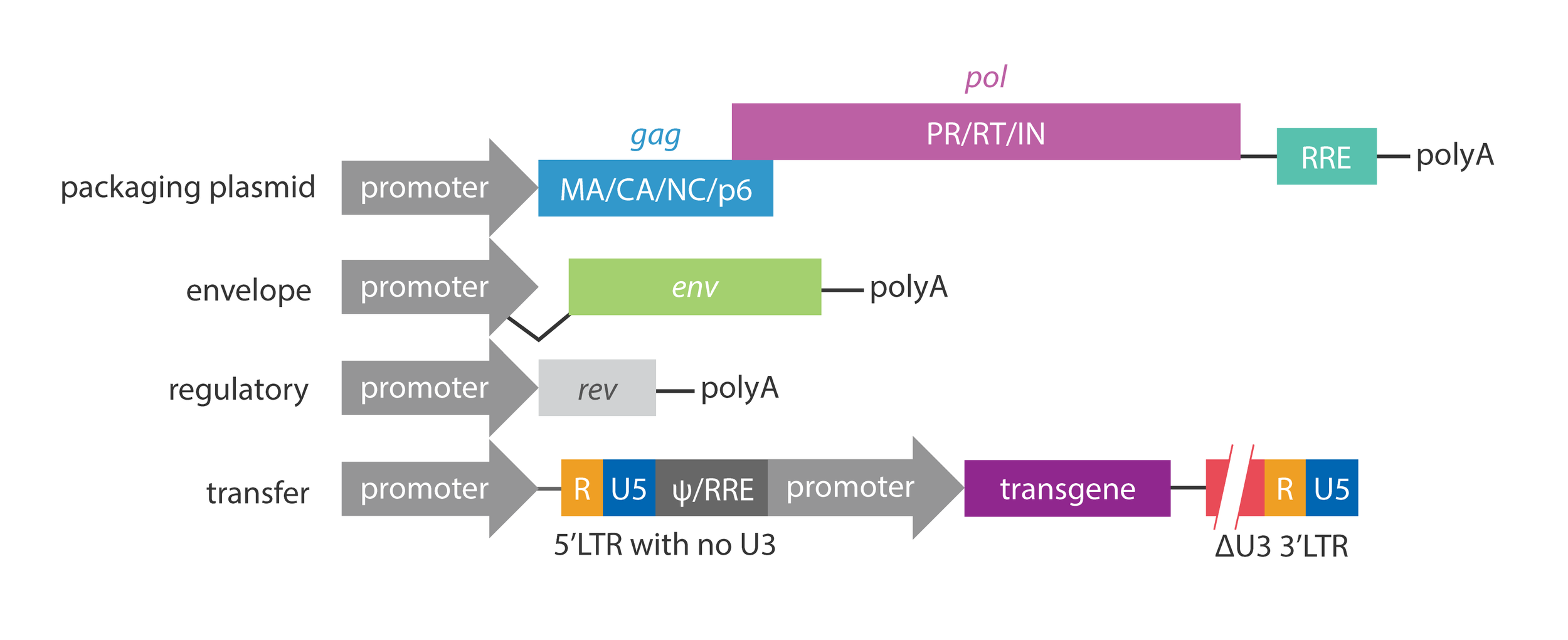
Modern approaches use a third-generation packaging system which splits critical lentiviral components across 4 separate plasmids to minimize the risk of the genetic elements recombining back into a complete, self-perpetuating virus. This 3rd gen. system also makes use of a self-inactivating (SIN) 3’ LTR to prevent mobilization/replication once the genetic material (aka “cargo” or “payload”) has been integrated into the target cell. Lastly, the 3rd gen. system uses a chimeric 5’ LTR to eliminate the need for TAT further minimizing viral sequence.
In wild type lentivirus, the Long Terminal Repeats (LTRs) are sequences on each end of the genome that play an essential role in many, if not all, steps of the viral life cycle. They are quite elegantly designed/evolved so I think it’s important to understand how they work and how they’ve been modified to serve science safely. Buckle up…
Each LTR consists of three parts: the U3 region (450nt), R region (100 nt), and U5 region (70 nt). After integrating into the host genome, the U3 acts as an enhancer/promoter that enables expression of the viral RNA transcript by recruiting host RNA polymerase II (RNAP II). It does this by containing multiple host factor binding site sequences including TF motifs for NF-KB and SP-1 which subsequently recruit host RNAP II. Downstream, the U3 also includes a TATA box. It’s everything you need to start transcription. The TSS of the lentiviral transcript is ~25 bp downstream of the TATA box, at the +1 position of the R region. Evidently, host RNAP II often stalls early in transcription of the viral genome (after 100nt or so) which is why the R region has evolved to, once transcribed, form a 5’ hairpin to recruit/bind the viral TAT (transactivator) protein to stabilize the transcription complex and greatly increase the # of viral RNAs produced. The R region also contains a “AAUAAA” polyA sequence that serves as the transcriptional stop signal for RNAP II once it reaches the other end of the integrated viral cassette.
Why doesn’t this polyA sequence pre-maturely terminate transcription since it’s also present in the R region of the 5’ LTR where transcription starts? B/c of U1 snRNP telescripting, a nuance of mammalian biology in which polyA stop sequences (which are actually quite common throughout the genome) are ignored by the transcription complex if it’s too close to an important part (exon) of a gene (determined by the presence of nearby splice junctions).
Before integration, the U5 region, along with the R region, serves as the anchor for the viral reverse transcriptase (RT) to initiate DNA synthesis off a bound host tRNA. These two regions also provide the grounds for the initial template switching event (see figure below) which is critical in converting the ssDNA into dsDNA for integration. Lastly, the outermost edges of the U3 and U5 regions (when arranged as proviral dsDNA) contains docking sites for the viral integrase (IN) protein to recognize, bind to, and ultimately clip (exposing hydroxyl groups that will perform a nucleophilic attack on host DNA) in order to integrate the provirus into the host genome. Focusing on the top strand of the dsDNA, the 5’ edge of the U3 contains the left attachment site (attL) whereas the 3’ edge of the U5 contains the right attachment (attR) site. These sites are inverted repeats (IRs) for reasons detailed below.

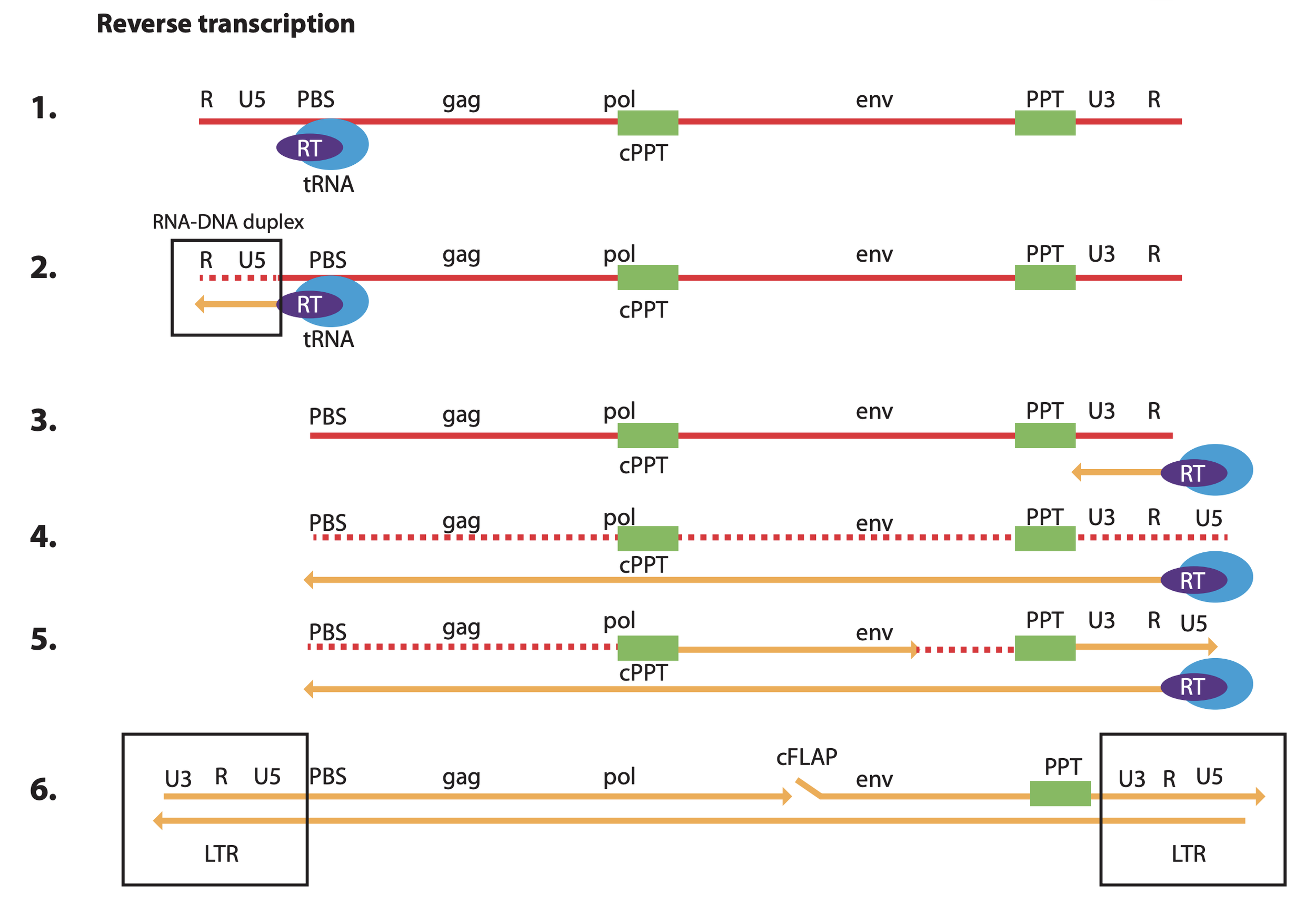
RNA is red w/ the initial seq. oriented 5’ to 3’. DNA is yellow. From https://www.nature.com/articles/s41375-018-0106-0#Fig1
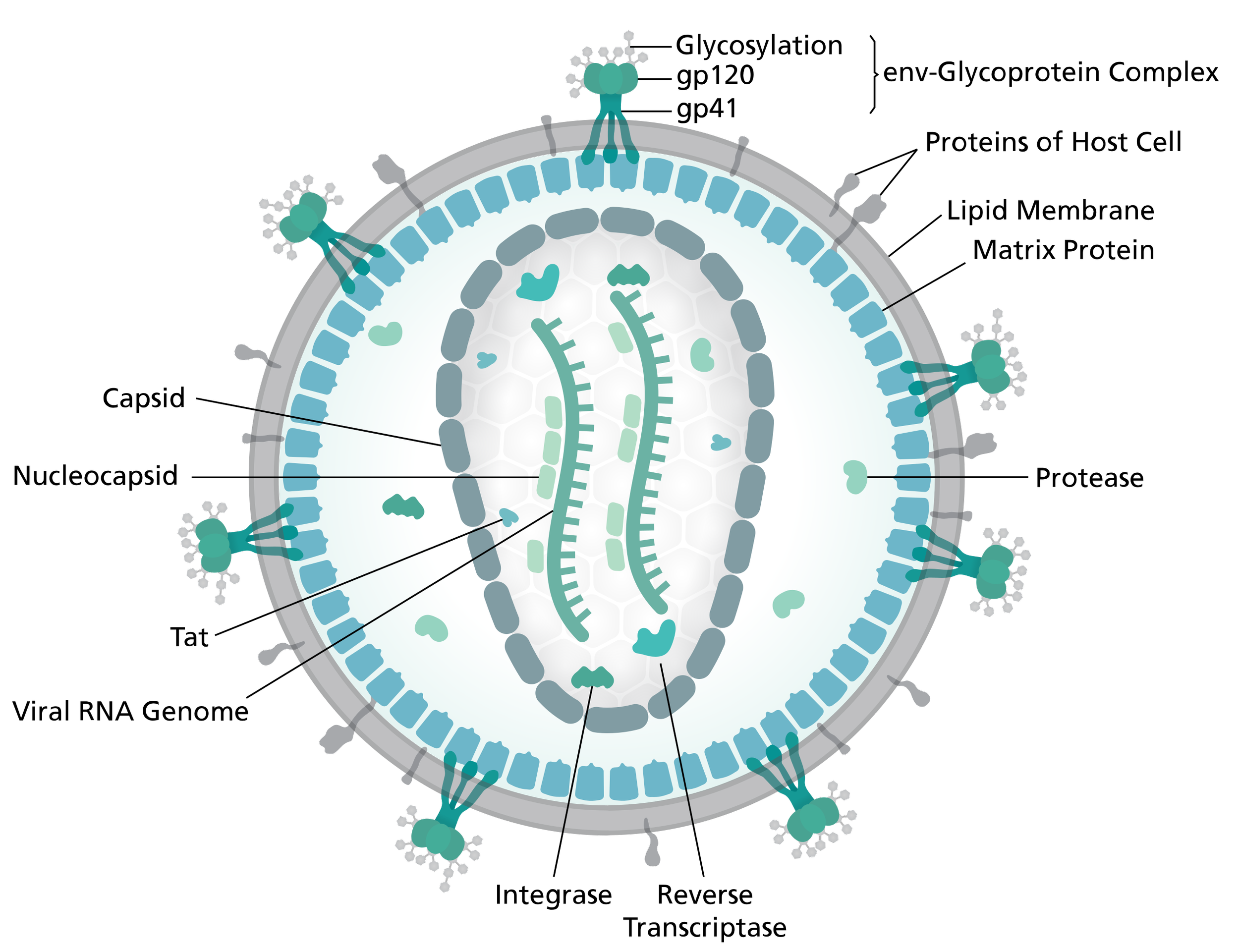
Lentivirus virion
By Thomas Splettstoesser (www.scistyle.com) through Wikimedia
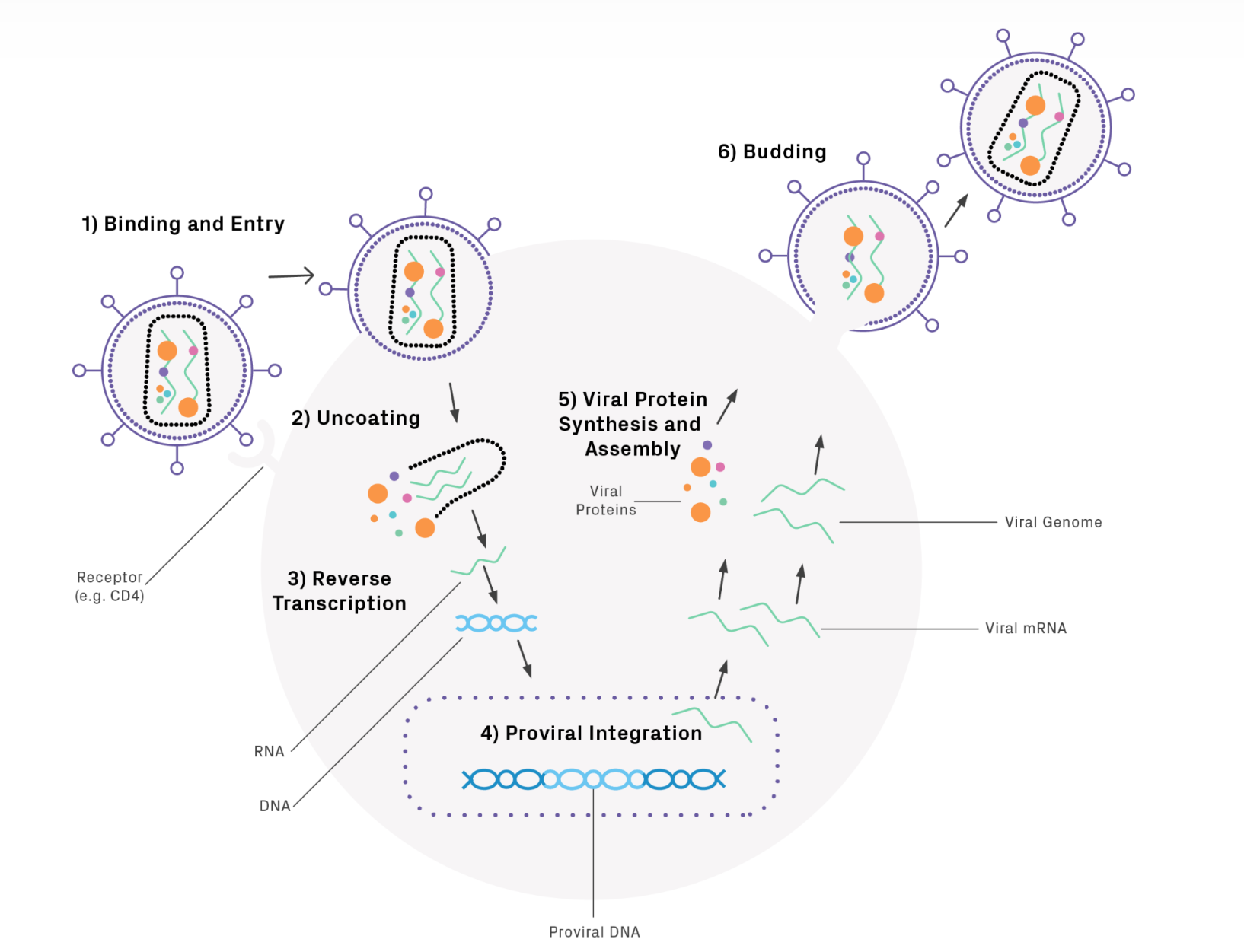
Lentivirus life cycle
Figure from Genewiz, the reverse transcription step is pretty wild as the virus uses a host tRNA to prime itself followed by a few template switching events to produce the “proviral” dsDNA which is then inserted into the host by the viral integrase (IN) protein. Note: retroviral reverse transcriptase (RT) has three core enzymatic functions that it uses sequentially: (1) RNA-dependent DNA polymerase, (2) ribonuclease H (RNase H), and (3) DNA-dependent DNA polymerase activity.
Inverted repeats on ends of proviral genome
A great figure summarizing what we know about lentivirus integration. Upon entering the cell, the positive sense ssRNA is primed by a host tRNA at the primer binding site (PBS). The viral RT then creates a complementary ssDNA copy of the U5 and R region while degrading the template RNA.
Remember: nucleic acid polymerization always occurs 5’ to 3’ in nature.
Next, the synthesized cDNA jumps (switches templates) to prime the other end of the viral RNA by R region complementarity. The viral RT is again recruited, this time copying the rest of the viral RNA into cDNA while degrading all of the orignal template EXCEPT for RNase H resistant polypurine tracts (PPT and cPPT).
The RNA remaining at the PPT sites primes synthesis of portions of the second DNA strand using the cDNA as template now. Why two separate initiation sites? Well you might notice the figure is missing something… how is the 5’ LTR ultimately reformed? Well, between steps (5) and (6) there is a second template switching step where the newly synthesized U3+R+U5(+PBS, not shown) DNA fragment created off the PPT site jumps back over to the left side of the forming provirus, binding the PBS complement on the bottom strand. Finally, with this last priming event, multiple copies of the viral RT complete the full dsDNA while creating a flap (cFLAP) that plays an important role in facilitating import of the provirus into the host nucelus.
In the end, it’s a beautifully evolved sequence that balances symmetry with directionality and functionality with compactness. Because the very ends of the proviral dsDNA genome are inverted repeats, the viral integrase only needs one docking site to work with either end, minimizing protein size and, thus, coding sequence (genomic “space”). However, the remainder of the LTRs are oriented as direct repeats to (1) enable cDNA synthesis via template switching and (2) maintain directionality during transcription. In the final proviral dsDNA that is incorporated into the host, the U3 is arranged to ensure only the coding sequences of the viral proteins are transcribed. If the full LTRs were arranged as inverted instead of direct repeats, both strands of the provirus would be transcribed from the outside in leading to a mess of collision events b/w different RNAP II complexes as well as anti-sense transcripts that inhibit one another and prevent viral protein synthesis.
Now, knowing all that, how do we modify lentivirus to safely work with while leveraging it’s capabilities? In general, the 3rd gen. system was designed to (1) produce large amounts of infectious particles containing nucleic acid sequences of our choosing that can integrate into the host genome (2) use as few parts / as little sequence from lentivirus as possible to minimize the risk of reassembly while simultaneously maximizing the amount of genomic space available for our own sequences of interest (3) ensure the virus is replication-incompetent once integrated (i.e. not transmissible) and (4) spread viral components across many plasmids (again, to minimize recombination) while still making the system easy to work with & reliable
The table above details the essential vs. optional lentiviral components for producing (achieving?) such a system. The figure below shows how the essential components are divided across separate plasmids. Importantly, we usually don’t use the lentiviral env gene (encoding the HIV envelope glycoprotein that binds CD4 on helper T cells) but instead swap it out with the env gene from Vesicular Stomatitis Virus (from the same family as Rabies virus) encoding a glycoprotein (VSV-G) that is much more broadly effective/infectious because it targets LDL receptors which are ubiquitously expressed on the surface of many different cell types. This is an example of a gain-of-function alteration of a virus that benefits research (shrug).
Perhaps most critical, and unique, to the 3rd gen. packaging system are the changes to the LTRs:
Truncation of 5' (chimeric) LTR: Makes the system Tat-independent. It allows the packaging cells to create the viral RNA genome using standard cellular machinery by fusing a strong human promoter to the start of the LTR in place of the U3 region. The R and U5 are kept to maintain transcription and integration capabilities.
Partial deletion of 3’ (SIN) LTR: Makes the virus Self-Inactivating. This "broken" 3' LTR containing a large deletion in the U3 region is copied to the 5' end during reverse transcription of the viral RNA into proviral dsDNA, ensuring that once the virus integrates into your target cell, the LTRs are “dead” and cannot produce new, full virus. Importantly, even though most of the U3 is deleted, the inverted repeat is maintain to enable integration.
Gemini goes into more detail which I’ve cleaned up to ensure accuracy…
The 5' LTR Modification: The "Tat-Independent" Chimera
In 3rd-generation plasmids, the 5' U3 region is replaced by a heterologous promoter (usually CMV or RSV).
The Modification: A truncation of the native 5' U3 and the insertion of a constitutive mammalian promoter.
The Goal: Tat-Independence. Wild-type HIV requires the Tat protein to stabilize transcription at the U3 region of the 5' LTR (which also contains NF-KB and SP1 motifs for TF binding and recruiting of RNA Pol II). By swapping U3 for CMV, the packaging cell (HEK293T) can churn out viral RNA without any Tat present.
The Safety Benefit: You can completely remove the tat gene from the packaging system. This reduces the number of viral genes used and minimizes the risk of recombinant competent retrovirus (RCR) forming.
The 3' LTR Modification: The "Self-Inactivating" (SIN) Design
The 3' LTR contains a significant deletion in its U3 region (often referred to as ΔU3) which is slightly different than the total truncation of the U3 region in the 5’ LTR
The Modification: Deletion of the enhancer and promoter elements within the 3' U3 sequence.
The Goal: Self-Inactivation. During reverse transcription in the target cell, the virus uses the 3' LTR as the template to build the new 5' LTR.
The Result: The "broken" 3' deletion is copied to the 5' end. The final integrated provirus ends up with no functional promoters at either end.
Why doesn’t modifying the LTRs prevent integration? The important end of each LTR, containing the integrase attachment (att) site, is preserved. The integrase enzyme sees a structurally sound genome with intact attL & attR site , allowing for high-efficiency integration, even though the U3 promoter sequences inside those LTRs are gone.
Altogether, this design cleverly creates a “one-way switch”
When you transfect the 3rd gen. transfer plasmid into 293Ts, the host cell's RNA Polymerase II sees that CMV promoter and starts transcribing the positive sense viral RNA
The Transcription Start Site (TSS): Crucially, the CMV promoter is positioned so that the transcript begins exactly at the start of the R region.
The Result: The cell produces a long strand of Full-Length Viral RNA. This RNA contains everything needed to be a virus: the (Psi) packaging signal, the Rev-Response Element (RRE), your transgene (for example, eGFP), and the 3' LTR. This is the "blueprint" that gets stuffed into the viral capsids.
Note: you should not include your own polyA stop signal as part of the sequence you want to introduce b/c it’s already built into the 3’ LTR and doing so would terminate the viral RNA transcript early preventing (or, at least, greatly inhibiting) successful packaging. Instead, we often include WPRE downstream of our transgene to stabilize the transcript as part of the 3’ UTR.
Once that virus infects your target cell, it converts its RNA back into DNA. As we discussed, during this conversion, the 3’ LTR (lacking a U3 but maintaining the integrase attachment sequence) is used as the template to rebuild the 5' LTR.
Now, look at the integrated provirus in your target cell:
The CMV is Gone: The CMV promoter from your original plasmid was upstream of the transcription start site. It was never part of the RNA, so it never made the trip to the target cell. It is left behind in the "trash" of the packaging cell.
The 5' U3 is Dead: In its place is the ΔU3 deletion copied from the 3' end. There is no promoter, no enhancer, and no "On" switch.
The Result: The host cell's RNA Polymerase looks at the beginning of the viral genome and sees nothing. It doesn't recognize it as a place to start transcription. Therefore, no full-length viral RNA can be made.
The Exception: Transgene expression off the internal promoter
If the LTRs are silent, how does our introduced transgene get expressed? This is why we include an internal promoter (like EF1a or PGK) as part of the cargo sequence.
The Strategy: This promoter is placed in the middle of the "cargo" area.
The Action: The cell can see this promoter. It starts transcribing right in the middle of the virus, reading only the eGFP sequence and stopping at the 3' LTR's polyA stop signal.
The Safety: Because this transcript starts after the (Psi) packaging signal, even if there were helper proteins around, this eGFP RNA wouldn’t be packaged into new virions.
Well, there you have it. The basics of how lentivirus works and how it’s been modified for science. From a deadly virus to an incredibly powerful tool for genomics research. Humans rule!
If you want more information…
Here’s a fantastic overview of lentivirus from Applied Biological Materials (ABM):
https://info.abmgood.com/lentivirus-system-introduction
And another one from Addgene:
The protocol below is for packaging lentivirus using the 3rd generation system (n=4 plasmids total) using constructs from the Trono lab available through Addgene
pGAG-POL: https://www.addgene.org/12251/
Weirdly named “pMDLg/pRRE”, I’ll refer to as pGAG-POL
pREV: https://www.addgene.org/12253/
Named “pRSV-Rev”, I’ll refer to as pREV
pVSV-G: https://www.addgene.org/12259/
Weirdly named “pMD2.G”, I’ll refer to as pVSV-G
Any 3rd gen compatible lenti transfer vector
This is the vector in which you insert your transgene(s) of interest. This vector changes from experiment-to-experiment unlike the other “structural” plasmids which remain constant and thus can be pre-mixed to simplify transfection step (see below).
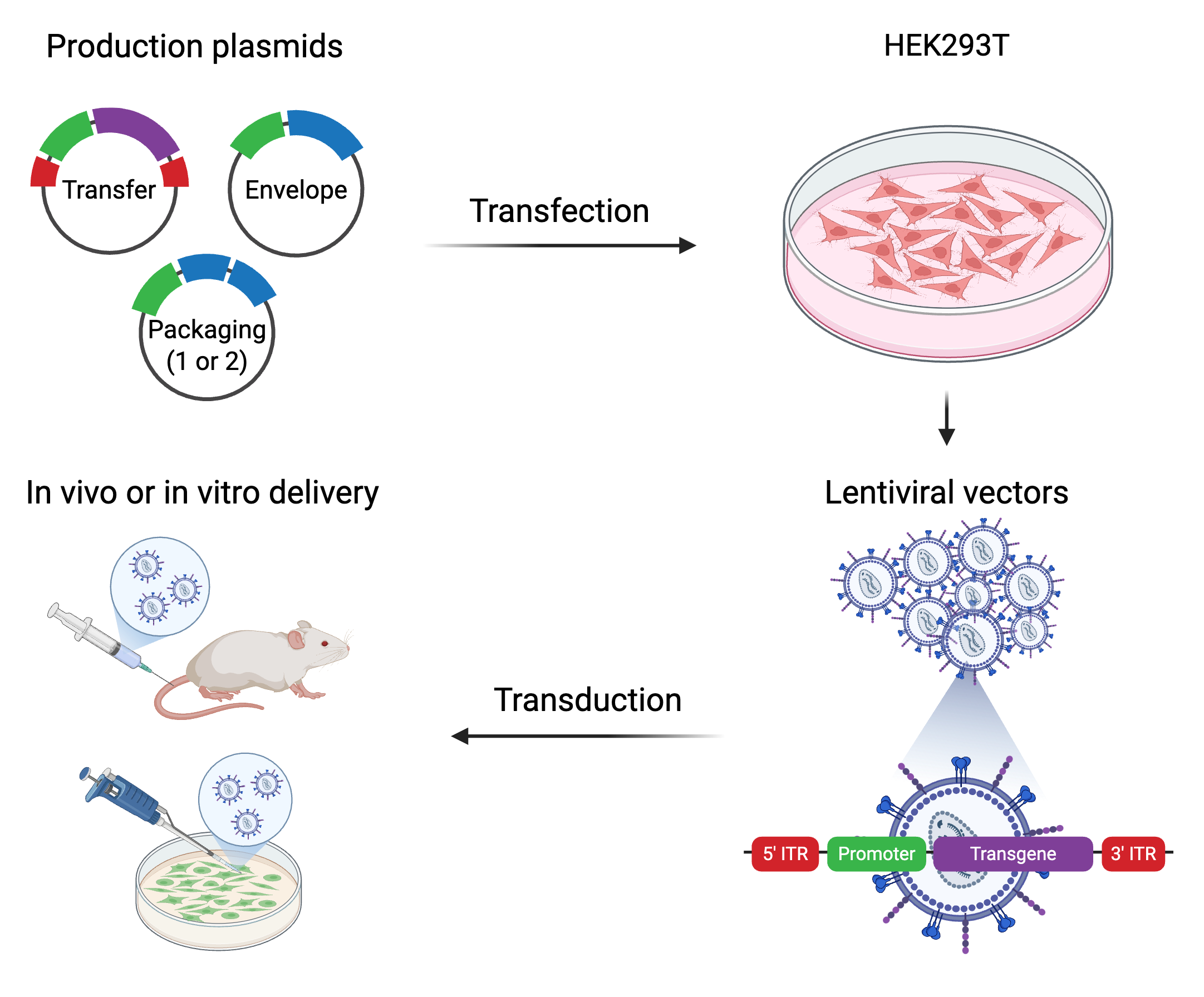
Example of 3rd gen. transfer plasmid: https://www.addgene.org/14883/
For this protocol, we’ll use a basic transfer vector expressing eGFP (with WPRE as part of 3’ UTR to enhance expression) driven by human polyubiquitin promoter-C (hUbC)
Components of this vector critical for lentivirus assembly include the 5’ and 3’ (truncated) LTRs, gp41 peptide, and Psi, RRE, cPPT/CTS elements
Weirdly named “FUGW”, I’ll refer to as pEGFP
High-titre, 3rd generation lentivirus packaging protocol
Achieves ~1E8 TUs/mL of standard pEGFP transfer vector when titered in 293Ts or K562s (TUs = IUs = transducible/infectious units of virus)
Minimum time to complete: 5-7 days
Material
1 vial of 5e6 Lenti-X (HEK)293T cells from Takara stored in liquid nitrogen (in cDMEM + 10% DMSO)
cDMEM i.e. Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS and “1X” penicillin (100 U/mL) & “1X” streptomycin (100 ug / mL)
Mirus Bio TransIT-LT1 transfection reagent… a proprietary mixture of cationic polymer (to bind DNA) and lipid coating to enable cellular uptake. “LT” = low-toxicity
Gibco Opti-MEM, pH 7.4 (reduced-serum medium) … recommended for use with cationic lipid transfection reagents
Takara Lenti-X Concentrator… a viscous mixture of PEG 6000 and salt in dPBS to bind/precipitate virions
Or make your own!
ALSTEM 500x ViralBoost reagent… to boost viral production in 293Ts, pretty sure it contains caffeine
Standard plasticware suitable for mammalian cell culture (e.g. treated plates, serological pipettes, etc.)
Purified & quantified lentiviral plasmids
pGAGPOL, pREV, and pVSVG, pre-mixed at a mass ratio of 1:1:2
Typically, I’ll make at a total concentration of 200 ng/uL (i.e. 50 ng pGAG-POL, 50 ng pREV, and 100 ng pVSVG per uL)
pEGFP (or any other lenti-based transfer vector compatible w/ the 3rd gen. packaging system)
4X viral concentator soln:
40% PEG 6000
1.2M NaCl
dPBS (pH = 7.4)
Mix 1 part concentrator soln with 3 parts (by vol.) cell medium containing lentivirus
Chill to 4C for 2-120h then spin 1500xg for 45-60 min in 4C refrigerated centrifuge

Preparing cells
(Day 0) Thaw 1 vial of lenti-X 293T cells (Takara) by retrieving from liquid nitrogen, immediately placing in sterile, 37C water bath for 2 min, then wiping down with paper towel sprayed w/ 70% EtOH and transferring to biosafety cabinet
Wash cells by transferring vial contents into 15 mL conical tube preloaded with 10 mL cDMEM. Spin diluted cells 300xg, 10 min. Resuspend cell pellet in 10 mL fresh cDMEM.
Transfer washed cells into 15cm dish preloaded with 21-23 mL warm cDMEM. Culture 44-48h undisturbed.
31-33 mL total volume in 15 cm dish. Initial cell concentration = 5e6 cells / 32 mL = 156,000 cells/mL
(Day 2) After 44-48h in culture, cells should be 70-90% confluent. Collect and count (e.g. with a Countess automated cell counter).
Remove & discard spent media, immediately add 10 mL of room temp. trypsin, incubate 5-10 min at 37C, then quench w/ 20-23 mL of warm cDMEM. Pipette to mix then transfer lifted cells to labeled 50 mL conical tube
Cells should total ~30e6. Passage by seeding 10x10cm dishes each with 2.5 – 3.0e6 cells total (in 11-13 mL of cDMEM per plate). Culture 18-24 hours.
If you have small amount of 293Ts leftover, dilute 1:100 into a 10cm or 15cm dish and incubate for 3-4 days. These cells will be used to titer the packaged virus.
(Day 3) After 18-24h, plates should be 50-70% confluent. This is the critical range of confluency for high yield/titer packaging.
Once confluency confirmed to be 50-70%, leave cells in incubator and move to benchtop to prepare DNA and lipid mixes separately, then combine to form transfection mixes:
Per sample (i.e. 10cm dish): Prepare DNA mix in ~1 mL total volume (using labeled 1.5 mL eppendorf tubes)
1 mL Opti-MEM
500 ng pGAGPOL
500 ng pREV
1000 ng pVSVG
8000 ng lentiviral plasmid
Mix by vortexing then spin down briefly
Note(1): I recommend a pre-mixed stock of pGAGPOL+pREV+pVSVG (“GRV” mix) at a 1:1:2 mass ratio and a total concentration = 200 ng/uL. Then, just add 10 uL of this mix per sample.
To prepare this pooled stock, since it’s so important and will be reused many times, I recommend maxiprepping each plasmid individually and verifying by whole plasmid (ONT) sequencing prior to mixing.
Note(2): For the transfer vector, a typical miniprep will return 25-40 uL of plasmid at 500-1000 ng/uL i.e. 10-40 ug total, enough for packaging n=1-5 dishes (10cm) worth of lentivirus
I.e. You’ll usually add 10-20 uL of transfer plasmid (500-1000 ng/uL) per sample
Separately, prepare the lipid mix
200 uL Opti-MEM / 10cm plate
30 uL Mirus TransIT-LT1 reagent / 10cm plate
I recommend preparing a master mix with 10-20% extra vol. in a 5-10mL tube
200 uL * 11 samples = 2200 uL of OptiMEM
30 uL * 11 samples = 330 uL of TransIT-LT1
Mix by pipetting up and down gently. DO NOT VORTEX OR SPIN DOWN.
Per 10cm plate, prepare final transfection mix (~1.2 mL total)
Pipette 220 uL of lipid master mix into each tube of prepared DNA mix without letting pipette touch side of tube then pipette gently to mix (do not vortex or spin down!).
Now ~1.2 mL total / tube
Allow final transfection mixes to sit at room temp. for 20-40 min for final DNA+lipid complexes to form. Do not vortex or spin down at any point!
Mirus recommends 15-30 min, I strongly recommend 10-20 min, timing is important here as the complexes grow with time and after a certain size (radius>400nm) they less efficiency get absorbed (endocytosed) by the cells
A great resource if you want to learn more: https://www.mirusbio.com/deliverance/?srsltid=AfmBOoq4JTbezLkY_BAdynufM1cQpWpusvzBNv1qfvK3XSCLMg4vhCis
Transfection, boosting, collection, and concentration
(Day 3): Once transfection mixes are created and given time for DNA+lipid to complex, add all 1.2ml of transfection mix dropwise to each 10cm dish of cells, swirling gently to distribute. Make sure to match plate ID w/ mix ID (and/or record paired IDs). Return cells to incubator.
Don’t need to change media a few hours after applying transfection mix b/c of it’s low toxicity (compared to Lipofectamine 3000)
(Day 4): 16-24h post transfection, add 22 uL of ALSTEM ViralBoost reagent (500x) per 10 cm plate. Swirl gently to mix then return cells to incubator.
Again, no change in media necessary. Let that virus accumulate!
(Day 5-6): 48h-72h post transfection, collect lentivirus. I usually collect the day after boosting, in the afternoon.
Transfer media (~11 mL / plate) from dish to 15 mL conical tube and centrifuge 500xg for 10 min (or 1000xg, 5 min) to pellet any cells/debris. Bleach & discard plates.
Pour clarified supernatant (containing lentivirus) into new, labeled 5-10cm TC plates for ease in next step. Bleach & discard tubes with pelleted cells/debris.
Use 10-20 mL syringe and attachable 0.45 CA filter to filter clarified supernatant into new, labeled 15 mL conical tube(s). Should recover ~11 mL of filtered sup. per sample
Bleach and discard plates that were holding the clarified sup. before it was filtered. I usually bleach and set aside to discard once I’m finished with everything else.
Add 3.5 mL of Takara lentiX concentrator (note: very viscous) per 11 mL of filtered lentivirus prep, mix by inversion, and incubate 4C for 4 - 48h
Yes, this is intentionally a very wide window. Packaged virus is actually stable at 4C in concentrator soln. for up to 1 week so choose based on your own schedule. I usually keep in fridge overnight unless I’m in a rush.
(Day 5-8): Once thoroughly chilled, centrifuge lentivirus preps 1500xg for 45 min at 4C using refrigerated, swinging bucket centrifuge compatible w/ 15 mL conical tubes.
Make sure to pre-chill centrifuge to 4C (takes 15-30 min). After centrifugation, you should see large, white pellet of precipitated virus at bottom of tubes.
Note: During the long centrifugation step is a good time to label your cryotubes for the final stage
Once virus is pelleted, remove supernatant from each tube by pouring out directly into 10% bleach (“decanting”). Be careful not have bleach backsplash into tube containing pellet.
After decanting all the tubes, carefully remove any remaining supernatant by vacuum aspiration or directly w/ pipette. I usually take extra care here to remove all residual liquid, at the expense of some precipitated virus.
Re-suspend white pellet (containing lentivirus) in 0.1X original volume (i.e. 1 mL) of dPBS. Mix well by pipetting.
Aliquot 330 uL of concentrated lentivirus into labeled cryotube then repeat (i.e. 3x330 ul aliquots per prep). Seal tightly and store at -80C indefinitely.
Big sigh of relief. Your construct is now packaged, concentrated, and saved.
Use concentrated lentivirus leftover in conical tubes (~10 uL / sample) to transduce control 293Ts (seeded 3-4 days ago at 0.01x dilution) to get an estimate of final yield/titers
Usually, I’ll collect the control 293Ts and measure then seed a 6-, 12-, or 24- well plate with 293Ts at a concentration of 1e5 cells / mL
Next, I add 1-10 uL of virus per well and swirl to distribute.
Lastly, bleach & discard tubes that previously contained pelleted virus and anything remaining that came in contact with lentivirus.
(Day 7-10): 44-56h post transduction, collect cells (at or nearing full confluency) and analyze by flow cytometry to measure %GFP+ of each population/sample
Note, this is only for the specific case in which you included a GFP expression cassette as part of your packaged sequence.
That’s it! You should now have an estimate of how well your lentivirus packaged and can proceed w/ transducing your cells of interest :)
Note: Estimates of viral titers in terms of transducible/infectious units are always relative as infectivity is highly dependent on cell type & mode of infection (e.g. simple mixing vs. spinfection). I usually do an initial test in 293Ts just to confirm our vectors were packaged and b/c it’s easy to set some cells aside during the packaging experiment but then test an aliquot of frozen virus again in my cell type of interest to get a more accurate estimate.
Feel free to scale the experiment as needed. I prefer packaging in 10cm plates but I’ve gone up to 15cm plates (for packaging lots of the same construct) and down to 96-well plates (for packaging many different constructs)
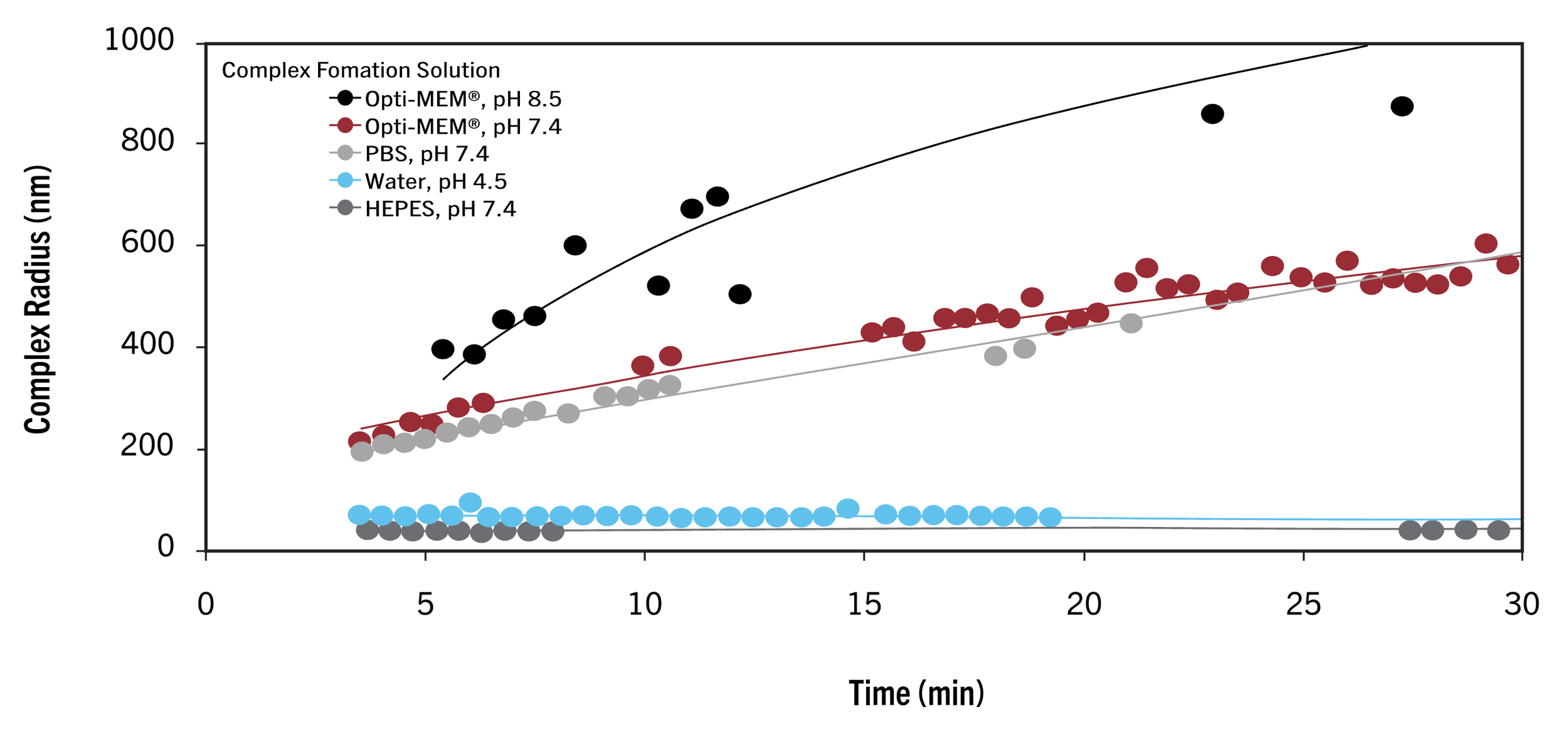
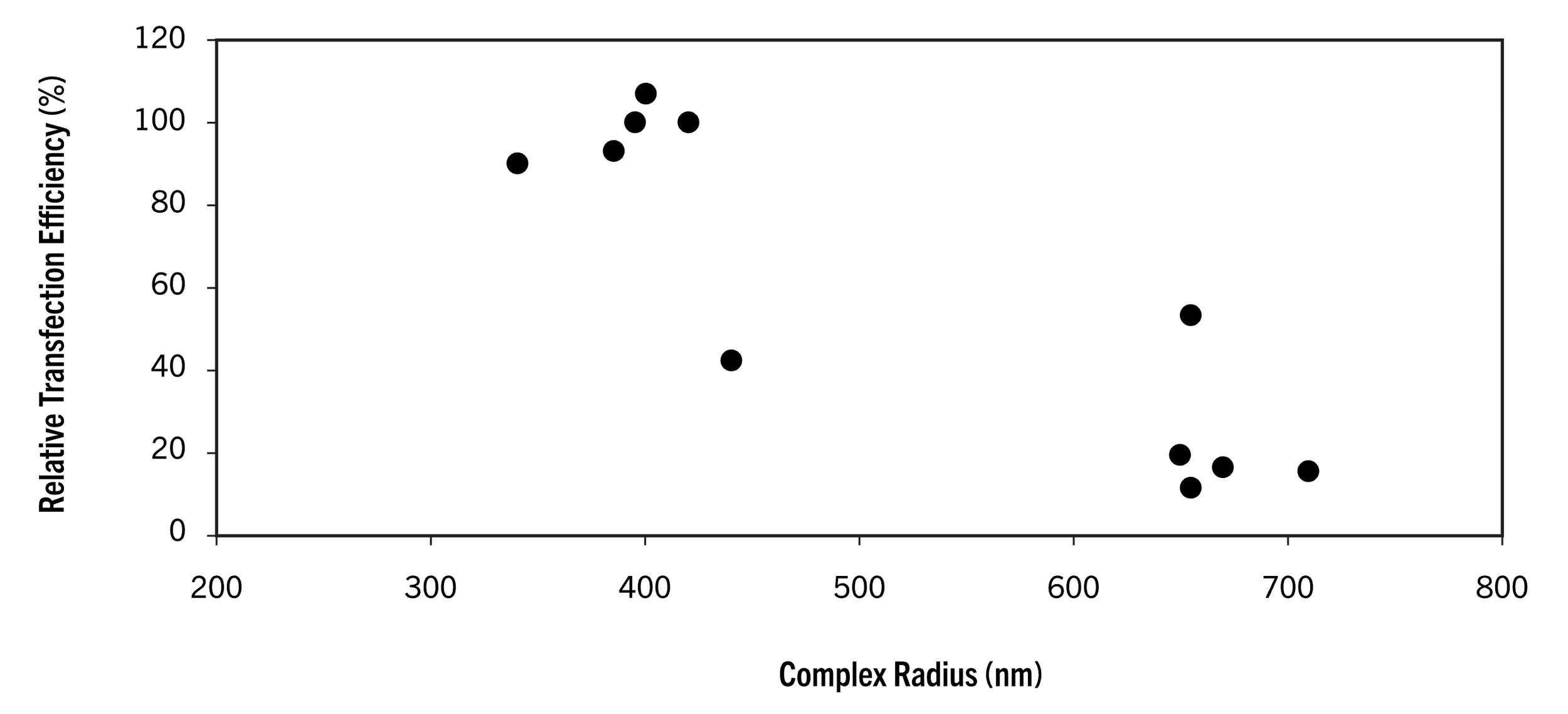
Based on Mirus’s data, using Opti-MEM (pH 7.4), we have ~15 min (once the lipid master mix has been combined w/ the DNA mix to form the final transfection mix) before the DNA+lipid complexes exceed a radius of 400 nm, at which point transfection efficiency greatly decreases. Now, I’m not sure which of their reagents their testing here, it’s possible the timing differs slightly for the TransIT-LT1 formula which I think includes a lipid and a cationic polymer, however their recommendation across protocols remains consistent: allow 10-30 min for DNA+lipid to complex then apply to cells.
Minimum time to complete packaging protocol: 5-7 days
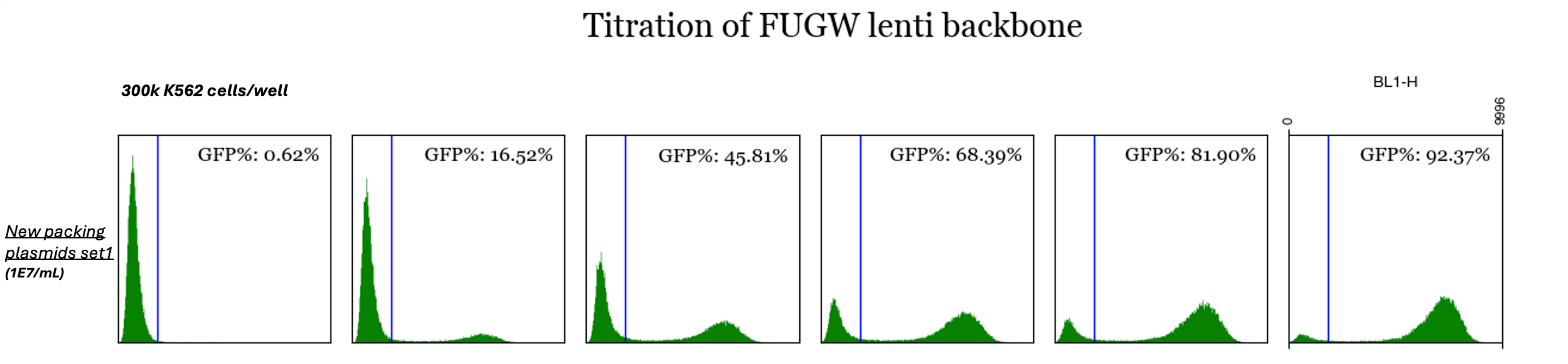
Data & figures from another fantastic graduate student in our lab (Weihao). Here, he achieved a packaged lentvirus titer of 1E7 TUs/mL in K562s w/o concentrating
the virus. He packaged pFUGW (“pEGFP”) in a 6-well plate (~2 mL/well) using 400 ng GRV plasmid mix + 1500 ng transfer plasmid per well and then applied
increasing amounts of the resulting virus to wells containing 300,000 cells and measured fluorescence after a couple days to estimate.
One last thing…
By definition of the genus, lentivirus include 2 copies of its +ssRNA genome in each virion. If we transfect 293Ts with a library of lentiviral transfer vectors, the odds are that the vast majority of packaged virions will contain two ssRNA encoding different constructs (CRISPR spacers) i.e. not exact copies (A+A) but heterodimers (A+B). This raises some immediate concerns…
Will each copy/construct lead to a different dsDNA provirus that integrates into the host and expresses separately? No. Fortunately for us, even though lentivirus contain 2 RNA copies, it almost always leads to 1 proviral integration event. Evolutionarily, the virus only needs to integrate once to persist and replicate so why do any extra work? But why then does it package 2 RNA copies? Because it helps with the complicated RT steps. Those template switching “jumps” we discussed earlier? Much more likely to occur w/ a 2nd copy present. The viral replication complex is complex with the RNA itself often folded over and/or containing a lot of secondary structure so if the RT gets stuck or falls off there’s a second copy there to latch on to and continue extending the cDNA. In general, the two copies appear to help one another in producing a single, full, dsDNA provirus. Therefore, if we control how many virions get inside each cell (by Poisson loading), we can ensure each cell incurs a single construct integration event (which is important in the interpretation of results).
If the 2 copies are helping each other out, doesn’t that mean they can produce a recombined product? Yes, and it’s something we need to keep in mind. Again, naturally, this is largely beneficial for the virus as it can handle any trouble during RT as well as evolve faster by combining the sequences of two slightly different RNAs (“strains”) during the RT step. These slightly different strains, which may separately carry beneficial (or deleterious) mutations, can either come from different virions that reached the host cell at a similar time or the same virion that was packaged w/ two slightly different genomes during it’s previous assembly (maybe some errors were introduced during the proviral transcription step in the last host before virions were assembled). This latter case most resembles what happens in our experiments. Even though we can control, roughly, how many virions get into the cell (transducing target cells at a low MOI to ensure each gets infected with 1 or 0 virions), there remains the high likelihood that the virion is a heterodimer and a non-zero possibility that these two “copies” recombine during the RT step to produce a new, chimeric, and unintended DNA sequence that gets integrated into the host. Now, the more sequence shared between the two RNA copies, the less likely for this to happen, which is why packaged CRISPR libraries are usually fine as the two RNA copies only differ by 20nt at one specific location in the middle of the RNA. However, if you’re packaging, for example, a cloned ORF library where each construct encodes a different protein or protein subunit, you’re gonna wanna be extra careful and check the level of recombination after the packaging step. Depending on how long your variable sequences are, this could be easy or difficult to do.